fgumi Best Practice FASTQ -> Consensus Pipeline
This document describes the recommended best practice pipeline for processing FASTQ files through to consensus sequences using fgumi.
Tools Required
This pipeline uses only fgumi and a read aligner:
- fgumi (version 0.1 or higher)
- bwa mem (version 0.7.17 or higher recommended)
Unlike fgbio-based pipelines, no samtools is required - fgumi provides native fastq, sort, and merge commands.
Common Configuration Options
Compression Level
fgumi supports compression levels 1-12 for BAM output:
| Use Case | Level | Notes |
|---|---|---|
| Final outputs | 6-9 | Balance of size and speed |
| Intermediate files | 1 | Fast compression, larger files |
| Piped commands | 1 | Minimize CPU overhead |
Set with --compression-level N on any command that writes BAM.
Threading
All major fgumi commands support multi-threading via --threads N:
# Single-threaded (default, optimized fast path)
fgumi group --input in.bam --output out.bam --strategy adjacency
# Multi-threaded with 8 threads
fgumi group --input in.bam --output out.bam --strategy adjacency --threads 8
Thread allocation is automatically optimized per-command based on workload profiling.
Memory
fgumi’s memory model differs significantly from fgbio’s JVM -Xmx. In particular, --queue-memory is per-thread by default and controls only pipeline queue backpressure — actual process memory will be higher. See the Performance Tuning Guide for detailed guidance, including a comparison table for fgbio users.
Boolean Flags
All boolean flags accept the following values (case-insensitive): true/false, yes/no,
y/n, t/f. For example:
fgumi filter --require-single-strand-agreement yes ...
fgumi simplex --output-per-base-tags true ...
fgumi group --allow-unmapped y ...
Pipeline Overview
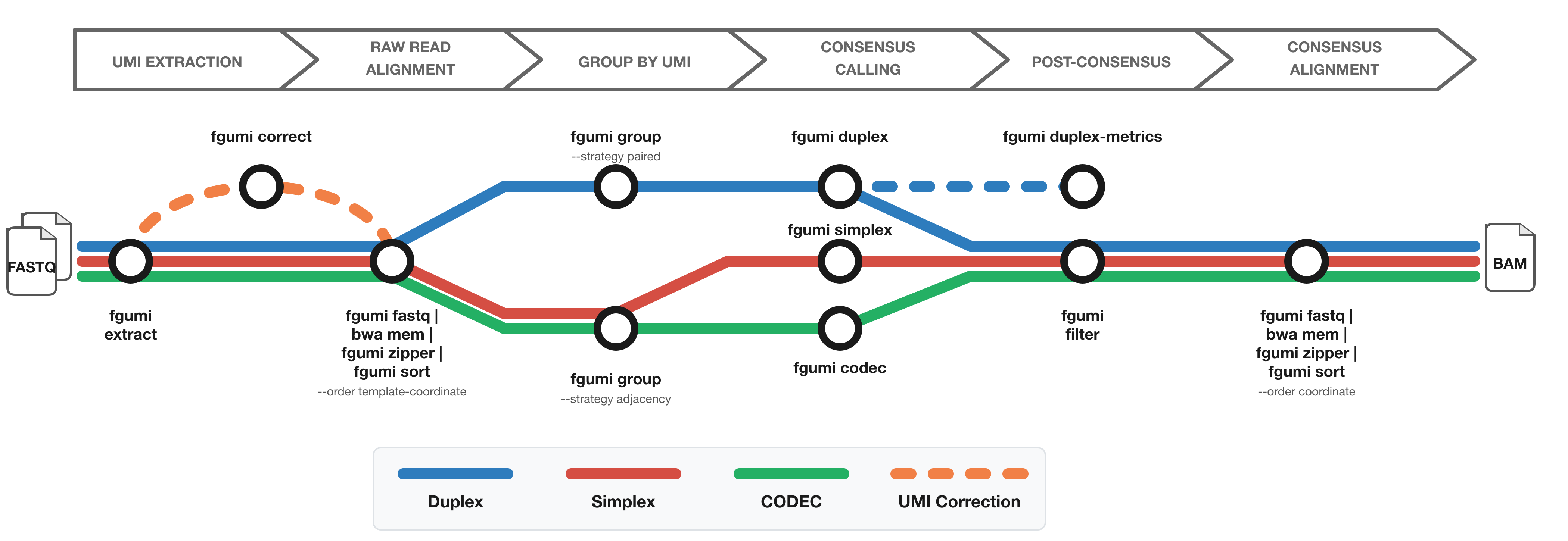
The diagram shows the workflow from FASTQ files to filtered consensus reads:
- Red: Simplex (single-strand) consensus
- Blue: Duplex (double-strand) consensus
- Green: CODEC consensus
- Orange: Optional UMI correction for fixed UMI sets
Phase 1: FASTQ → Grouped BAM
graph TD;
A["fgumi extract"]-->B["fgumi fastq | bwa mem | fgumi zipper"];
B-->C["fgumi sort"];
C-->D["fgumi merge (optional)"];
D-->E["fgumi group"];
Phase 2a: Grouped BAM → Filtered Consensus (R&D Version)
graph TD;
A["fgumi simplex/duplex"]-->B["fgumi fastq | bwa mem | fgumi zipper"];
B-->C["fgumi filter | fgumi sort"];
Phase 2b: Aligned BAM → Filtered Consensus (High-Throughput Version)
graph TD;
A["fgumi simplex/duplex"]-->B["fgumi fastq | bwa mem | fgumi zipper | fgumi filter | fgumi sort"];
Phase 1: FASTQ to Grouped BAM
Step 1.1: UMI Extraction
Convert FASTQ files to unmapped BAM with UMI extraction:
fgumi extract \
--inputs r1.fq.gz r2.fq.gz \
--read-structures 8M+T +T \
--sample "sample_name" \
--library "library_name" \
--output unmapped.bam \
--threads 4
Key parameters:
--read-structures: Define UMI and template positions (e.g.,8M+T= 8bp UMI + template)
For dual-index UMIs (duplex sequencing), use paired read structures:
fgumi extract \
--inputs r1.fq.gz r2.fq.gz \
--read-structures 8M+T 8M+T \
--sample "sample_name" \
--library "library_name" \
--output unmapped.bam
Optional: UMI Error Correction
For fixed/known UMI sets, correct sequencing errors before alignment:
fgumi correct \
--input unmapped.bam \
--output corrected.bam \
--umi-files known_umis.txt \
--min-distance 1
Step 1.2: Alignment
Align reads using the fgumi fastq + zipper pipeline:
fgumi fastq --input unmapped.bam \
| bwa mem -t 16 -p -K 150000000 -Y ref.fa - \
| fgumi zipper --unmapped unmapped.bam --reference ref.fa --output aligned.bam
Key points:
fgumi fastqconverts BAM to interleaved FASTQ for the aligner-ptells bwa mem to expect interleaved paired-end reads-K 150000000sets batch size (improves reproducibility)-Yis critical: Use soft-clipping for supplementary alignments to preserve basesfgumi zippertransfers tags from unmapped BAM to aligned readsfgumi zipperaccepts SAM or BAM on stdin or--input. For best performance, pipe uncompressed BAM from the aligner (e.g.bwa-mem3 mem --bam=0); SAM is fine for aligners that can’t emit BAM
For large files, add threading:
fgumi fastq --input unmapped.bam --threads 4 \
| bwa mem -t 16 -p -K 150000000 -Y ref.fa - \
| fgumi zipper --unmapped unmapped.bam --reference ref.fa --output aligned.bam --threads 4
Step 1.3: Sorting
Sort into template-coordinate order before grouping:
fgumi sort \
--input aligned.bam \
--output sorted.bam \
--order template-coordinate \
--threads 8 \
--max-memory 4G
For single-cell data, the CB cell barcode tag is automatically included in the
template-coordinate sort key, keeping templates from different cells at the same locus separate:
fgumi sort \
--input aligned.bam \
--output sorted.bam \
--order template-coordinate \
--threads 8
Step 1.3b: (Optional) Merging Multiple BAMs
When processing multiple lanes or flowcells separately, merge the sorted BAMs before grouping.
fgumi merge performs an efficient k-way merge without re-sorting:
fgumi merge \
--order template-coordinate \
--output merged.bam \
lane1_sorted.bam lane2_sorted.bam lane3_sorted.bam
For large numbers of files, use --input-list:
fgumi merge \
--order template-coordinate \
--input-list bam_paths.txt \
--output merged.bam
For single-cell data, the CB cell barcode tag is automatically included in the merge key.
All inputs must be sorted in the same order as --order. Do not use samtools merge for
template-coordinate BAMs — it does not understand the tc tag that fgumi zipper adds, and
will produce incorrect ordering.
Step 1.4: UMI Grouping
Group reads by UMI using the appropriate strategy:
For simplex/single-UMI workflows:
fgumi group \
--input sorted.bam \
--output grouped.bam \
--strategy adjacency \
--edits 1 \
--metrics group_metrics \
--threads 8
For duplex/paired-UMI workflows:
fgumi group \
--input sorted.bam \
--output grouped.bam \
--strategy paired \
--edits 1 \
--metrics group_metrics \
--threads 8
The --metrics PREFIX flag writes all three metrics files in one step:
PREFIX.family_sizes.txt— family size histogramPREFIX.grouping_metrics.txt— grouping statisticsPREFIX.position_group_sizes.txt— UMI families per genomic position
These can also be written to explicit paths with --family-size-histogram and
--grouping-metrics.
For workflows with unmapped templates (e.g., some cfDNA assays):
fgumi group \
--input sorted.bam \
--output grouped.bam \
--strategy adjacency \
--allow-unmapped \
--metrics group_metrics
By default, templates where all reads are unmapped are excluded. --allow-unmapped includes
them so their UMIs are still tracked and grouped with any mapped reads from the same molecule.
Step 1.5: (Optional) QC Metrics Before Consensus
For simplex libraries, collect QC metrics from the grouped BAM:
fgumi simplex-metrics \
--input grouped.bam \
--output simplex_metrics \
--min-reads 3
This produces simplex_metrics.family_sizes.txt, simplex_metrics.simplex_yield_metrics.txt,
simplex_metrics.umi_counts.txt, and optionally a PDF plot. The yield metrics show how the
number of callable consensus reads scales with sequencing depth (computed at 5%, 10%, …, 100%
of reads), so you can assess whether deeper sequencing would materially improve yield.
For duplex libraries, use duplex-metrics:
fgumi duplex-metrics \
--input grouped.bam \
--output duplex_metrics
Phase 2a: R&D Pipeline (Separate Consensus and Filtering)
This approach generates an intermediate consensus BAM, allowing you to experiment with different filtering parameters without re-running consensus calling.
Step 2a.1: Consensus Calling
Simplex consensus:
fgumi simplex \
--input grouped.bam \
--output consensus.bam \
--min-reads 1 \
--min-input-base-quality 20 \
--output-per-base-tags true \
--threads 8
Duplex consensus:
fgumi duplex \
--input grouped.bam \
--output consensus.bam \
--min-reads 1 \
--min-input-base-quality 20 \
--output-per-base-tags true \
--threads 8
Key parameters:
--min-reads 1: Keep all consensus reads (filter later)--output-per-base-tags true: Enable per-base filtering downstream--min-input-base-quality: Minimum quality for input bases (default: 10)
Note: --output-per-base-tags accepts true/false, yes/no, y/n, or t/f.
Step 2a.2: Re-alignment
Consensus reads are unmapped and must be re-aligned:
fgumi fastq --input consensus.bam \
| bwa mem -t 16 -p -K 150000000 -Y ref.fa - \
| fgumi zipper --unmapped consensus.bam --reference ref.fa --output consensus.mapped.bam
Step 2a.3: Filtering
Filter consensus reads with desired stringency:
Simplex filtering:
fgumi filter \
--input consensus.mapped.bam \
--output filtered.bam \
--ref ref.fa \
--min-reads 3 \
--max-read-error-rate 0.025 \
--max-base-error-rate 0.1 \
--min-base-quality 40 \
--max-no-call-fraction 0.2 \
--reverse-per-base-tags \
--threads 8
Duplex filtering (with strand-specific thresholds):
fgumi filter \
--input consensus.mapped.bam \
--output filtered.bam \
--ref ref.fa \
--min-reads 10,5,3 \
--max-read-error-rate 0.025 \
--max-base-error-rate 0.1 \
--min-base-quality 40 \
--max-no-call-fraction 0.2 \
--reverse-per-base-tags \
--require-single-strand-agreement true \
--threads 8
For duplex, --min-reads 10,5,3 means:
- 10 raw reads minimum for final duplex consensus
- 5 raw reads minimum for AB single-strand consensus
- 3 raw reads minimum for BA single-strand consensus
Step 2a.4: Final Sort (if needed)
Sort to coordinate order for downstream tools:
fgumi sort \
--input filtered.bam \
--output final.bam \
--order coordinate \
--threads 8
Phase 2b: Aligned BAM → Filtered Consensus (High-Throughput Version)
For production use where filtering parameters are established, combine steps for better throughput.
Stage 1: Group and call consensus in a single pipe:
fgumi group --input aligned.bam --strategy adjacency --threads 4 --compression-level 1 \
| fgumi simplex --input /dev/stdin --min-reads 1 --output-per-base-tags true \
--output consensus.bam --threads 4 --compression-level 1
Stage 2: Align, filter, and sort in a single pipe:
fgumi fastq --input consensus.bam \
| bwa mem -t 16 -p -K 150000000 -Y ref.fa - \
| fgumi zipper --unmapped consensus.bam --reference ref.fa \
| fgumi filter --input /dev/stdin --ref ref.fa --min-reads 3 \
| fgumi sort --input /dev/stdin --output filtered.bam --order coordinate --threads 4
Note: The two stages cannot be combined into a single pipeline because fgumi zipper --unmapped needs random access to the consensus BAM. For most use cases, the R&D pipeline with intermediate files provides better debuggability and flexibility.
Alternative: Deduplication Without Consensus
For workflows that need UMI-aware duplicate marking without consensus calling (e.g., when downstream tools handle deduplication differently, or for QC purposes), use fgumi dedup:
graph TD;
A["fgumi extract"]-->B["fgumi fastq | bwa mem | fgumi zipper"];
B-->C["fgumi sort --order template-coordinate"];
C-->D["fgumi dedup"];
Dedup Pipeline
# Step 1: Extract UMIs from FASTQ
fgumi extract \
--inputs r1.fq.gz r2.fq.gz \
--read-structures 8M+T 8M+T \
--sample "sample_name" \
--library "library_name" \
--output unmapped.bam
# Step 2: Align reads (fgumi zipper adds required `tc` tag)
fgumi fastq --input unmapped.bam \
| bwa mem -t 16 -p -K 150000000 -Y ref.fa - \
| fgumi zipper --unmapped unmapped.bam --reference ref.fa --output aligned.bam
# Step 3: Sort with fgumi (required - samtools sort won't work)
fgumi sort --input aligned.bam --output sorted.bam --order template-coordinate
# Step 4: Mark duplicates
fgumi dedup --input sorted.bam --output deduped.bam --metrics metrics.txt
Important: You MUST use fgumi zipper and fgumi sort before fgumi dedup:
fgumi zipperadds thetc(template-coordinate) tag to secondary/supplementary readsfgumi sort --order template-coordinatekeeps all alignments for a template together; downstreamfgumi dedupuses thetctag to validate inputsamtools sort --template-coordinatedoes NOT understand thetctag and will produce incorrect results for dedup
Dedup Options
# Remove duplicates instead of marking
fgumi dedup --input sorted.bam --output deduped.bam --remove-duplicates true
# Use a different UMI strategy (default: adjacency)
fgumi dedup --input sorted.bam --output deduped.bam --strategy paired --edits 1
# Write family size histogram
fgumi dedup --input sorted.bam --output deduped.bam \
--metrics metrics.txt \
--family-size-histogram histogram.txt
Recommended Parameters by Application
Variant Calling (High Sensitivity)
fgumi simplex --min-reads 1 --min-input-base-quality 10
fgumi filter --min-reads 2 --max-base-error-rate 0.2 --max-no-call-fraction 0.3
Variant Calling (High Specificity)
fgumi duplex --min-reads 1 --min-input-base-quality 20
fgumi filter --min-reads 10,5,3 --max-base-error-rate 0.1 --max-no-call-fraction 0.1 \
--require-single-strand-agreement true
Liquid Biopsy / ctDNA
fgumi duplex --min-reads 1 --min-input-base-quality 20
fgumi filter --min-reads 3,2,2 --max-base-error-rate 0.05 \
--require-single-strand-agreement true
Troubleshooting
Low Consensus Yield
- Check family size distribution with
--metricsonfgumi group - Lower
--min-readsthreshold - Verify UMI extraction with correct
--read-structures - Run
fgumi simplex-metricsorfgumi duplex-metricson the grouped BAM to assess yield curves
High Error Rates
- Increase
--min-input-base-qualityduring consensus calling - Tighten
--max-base-error-rateduring filtering - For duplex, use
--require-single-strand-agreement true
Memory Issues
- Use
--max-memorywithfgumi sortto limit RAM usage - Reduce
--threads(fewer threads = less memory) - Process in smaller batches
- See Performance Tuning for detailed guidance
See Also
- UMI Grouping — grouping strategies and cell barcode support
- Working with Metrics — metrics file formats and interpretation
- Performance Tuning — threading, memory, and compression