Home
fgumi
High-performance tools for UMI-tagged sequencing data: extraction, grouping, and consensus calling.
The diagram shows the workflow from FASTQ files to filtered consensus reads:
- Red: Simplex (single-strand) consensus
- Blue: Duplex (double-strand) consensus
- Green: CODEC consensus
- Orange: Optional UMI correction for fixed UMI sets
Where to Use fgumi
Command Line
Install and run fgumi directly on your data. See the Getting Started guide.
Nextflow Pipeline
Use fastquorum for an end-to-end Nextflow workflow from FASTQ to consensus reads using fgumi.
Latch.bio
Run fgumi in the cloud with a point-and-click interface via Latch.bio — no installation required.
Installation
Pre-built Binaries
Pre-built binaries for common operating systems and architectures are attached to each release.
Cargo
cargo install fgumi
Bioconda
conda install -c bioconda fgumi
From Source
git clone https://github.com/fulcrumgenomics/fgumi
cd fgumi
cargo build --release
Available Commands
| Command | Description |
|---|---|
extract | Extract UMIs from FASTQ files |
correct | Correct UMIs based on sequence similarity |
fastq | Convert BAM to FASTQ format |
zipper | Restore original FASTQ from unaligned BAM |
sort | Sort BAM by coordinate/queryname/template |
group | Group reads by UMI |
dedup | Mark/remove UMI-aware duplicates |
simplex | Call single-strand consensus reads |
duplex | Call duplex consensus reads |
codec | Call CODEC consensus |
filter | Filter consensus reads |
clip | Clip overlapping read pairs |
duplex-metrics | Collect duplex metrics |
review | Review consensus variants |
downsample | Downsample BAM by UMI family |
simplex-metrics | Collect simplex metrics |
merge | Merge sorted BAM files |
See the Tool Reference for detailed documentation of each command.
Getting Started
This guide walks through a basic fgumi workflow from FASTQ files to filtered consensus reads.
Prerequisites
- fgumi installed (see Installation)
- A reference genome FASTA (with BWA index)
- Paired-end FASTQ files with UMI sequences
Basic Workflow
1. Extract UMIs from FASTQ
Extract UMIs from FASTQ reads and create an unmapped BAM. The --read-structures argument tells fgumi where UMI bases are located in each read. See Read Structures for details.
fgumi extract \
--inputs R1.fastq.gz R2.fastq.gz \
--read-structures +T +M \
--output unaligned.bam \
--sample MySample \
--library MyLibrary
2. (Optional) Correct UMIs
If using a fixed set of known UMIs, correct sequencing errors:
fgumi correct \
--input unaligned.bam \
--output corrected.bam \
--umi-files umis.txt \
--min-distance 1
3. Align and Sort
Use fgumi’s streaming pipeline to align with BWA and sort into template-coordinate order in a single pass:
fgumi fastq --input unaligned.bam \
| bwa mem -p ref.fa - \
| fgumi zipper --unmapped unaligned.bam \
| fgumi sort --output sorted.bam --order template-coordinate
This pipes reads through:
fastq— converts unmapped BAM to interleaved FASTQbwa mem— aligns reads to the referencezipper— merges aligned reads with original unmapped BAM to restore UMI tagssort— sorts into template-coordinate order for grouping
Note:
fgumi zipperaccepts SAM or BAM input, on stdin or via--input. For best performance, pipe uncompressed BAM from the aligner (e.g.bwa-mem3 mem --bam=0) — this skips both the SAM text formatting on the aligner side and the SAM parsing on the zipper side. SAM is fine for aligners that can’t emit BAM; compressed BAM on a pipe is not recommended (wasted CPU on both ends).
For single-cell data, the CB cell barcode tag is automatically included in the
template-coordinate sort key, keeping templates from different cells at the same locus separate:
fgumi fastq --input unaligned.bam \
| bwa mem -p ref.fa - \
| fgumi zipper --unmapped unaligned.bam \
| fgumi sort --output sorted.bam --order template-coordinate
3b. (Optional) Merge Multiple BAMs
If processing multiple lanes or flowcells separately, merge the sorted BAMs before grouping:
fgumi merge \
--order template-coordinate \
--output merged.bam \
lane1_sorted.bam lane2_sorted.bam lane3_sorted.bam
All inputs must be sorted in the same order. For large numbers of files, use --input-list:
fgumi merge \
--order template-coordinate \
--input-list bam_paths.txt \
--output merged.bam
For single-cell data, the CB cell barcode tag is automatically included in the merge key.
4. Group Reads by UMI
Group reads from the same original molecule together.
For duplex workflows, use paired strategy:
fgumi group \
--input sorted.bam \
--output grouped.bam \
--strategy paired
For simplex/codec workflows, use adjacency strategy:
fgumi group \
--input sorted.bam \
--output grouped.bam \
--strategy adjacency
To collect all grouping QC metrics under a single prefix:
fgumi group \
--input sorted.bam \
--output grouped.bam \
--strategy adjacency \
--metrics group_metrics
This writes group_metrics.family_sizes.txt, group_metrics.grouping_metrics.txt, and
group_metrics.position_group_sizes.txt in one step.
See UMI Grouping for details on grouping strategies.
5. Call Consensus Reads
Choose the consensus calling method based on your library preparation:
Simplex consensus (single-strand):
fgumi simplex \
--input grouped.bam \
--output consensus.bam
Duplex consensus (double-strand):
fgumi duplex \
--input grouped.bam \
--output duplex.bam
CODEC consensus:
fgumi codec \
--input grouped.bam \
--output codec_consensus.bam
See Consensus Calling and Duplex Consensus Calling for details.
6. (Optional) Collect QC Metrics
Collect QC metrics before filtering to understand your library.
For simplex libraries, use simplex-metrics on the grouped BAM:
fgumi simplex-metrics \
--input grouped.bam \
--output simplex_metrics
For duplex libraries, use duplex-metrics on the grouped BAM:
fgumi duplex-metrics \
--input grouped.bam \
--output duplex_metrics
Both commands write a set of metrics files under the given output prefix. See Working with Metrics for details on interpreting the output.
7. Filter Consensus Reads
Filter consensus reads based on quality metrics. The --min-reads format depends on the
consensus type:
For simplex consensus (single integer):
fgumi filter \
--input consensus.bam \
--output filtered.bam \
--ref ref.fa \
--min-reads 1
For duplex consensus (three comma-separated values: duplex,AB,BA):
fgumi filter \
--input duplex.bam \
--output filtered.bam \
--ref ref.fa \
--min-reads 1,1,1
8. (Optional) Clip Overlapping Reads
Clip overlapping bases in read pairs to avoid double-counting evidence:
fgumi clip \
--input filtered.bam \
--output clipped.bam \
--ref ref.fa
What’s Next
- Best Practices — recommended parameter settings and pipeline configuration
- Performance Tuning — threading, memory, and compression optimization
- Working with Metrics — understanding fgumi’s output metrics
Read Structures
Overview
A Read Structure is a string that describes how the bases in a sequencing run should be allocated into logical reads. It serves a similar purpose to the --use-bases-mask in Illumina’s bcl-convert, but provides additional capabilities.
A Read Structure is a sequence of <number><operator> pairs (called segments). The last segment may use + instead of a number to mean “whatever bases remain.” fgumi uses the read-structure crate for parsing and validation.
Read structures are used primarily in fgumi extract to specify where UMI bases, template bases, and other sequences are located in each FASTQ read.
Operators
Five kinds of operator are supported:
| Operator | Name | Meaning |
|---|---|---|
T | Template | Reads of template (e.g. genomic DNA, RNA) |
B | Sample Barcode | Index sequence for sample identification |
M | Molecular Barcode | UMI sequence for identifying the source molecule |
C | Cell Barcode | Index sequence for identifying the cell (single-cell) |
S | Skip | Bases to skip or ignore (e.g. monotemplate from library prep) |
Rules
- Any number of segments >= 1 is valid
- The length of each segment must be a positive integer >= 1, or
+ - Only the last segment in a read structure may use
+for its length - Adjacent segments may use the same operator (e.g.
6B6B+Tis valid if two sample indices are ligated separately)
Examples
Simple paired-end (2x150bp, no indices)
Per-read structures: +T, +T
Paired-end with 8bp sample index
Per-read structures: +T, 8B, +T
Paired-end with inline 6bp UMI in R1
Per-read structures: 6M+T, 8B, +T
The first 6 bases of R1 are the UMI, followed by template.
Duplex sequencing with dual barcoding and UMI + monotemplate
Per-read structures: 10M5S+T, 8B, 8B, 10M5S+T
Both R1 and R2 start with a 10bp UMI followed by 5bp of monotemplate (skipped), then template.
Single-cell with cell barcodes and UMI
Per-read structures: 5C30S5C3S8M+T, 8B, +T
R1 contains two cell barcodes separated by linker sequences, then a UMI, then template.
Formal Grammar
<read-structure> ::= <fixed-structure> <segment>
<fixed-structure> ::= "" | <fixed-length> <operator> <fixed-structure>
<segment> ::= <fixed-length> <operator> | <variable-length> <operator>
<operator> ::= "T" | "B" | "M" | "C" | "S"
<fixed-length> ::= <non-zero-digit>{<digit>}
<variable-length> ::= "+"
<any-length> ::= <fixed-length> | <variable-length>
<non-zero-digit> ::= "1" | "2" | "3" | "4" | "5" | "6" | "7" | "8" | "9"
<digit> ::= "0" | <non-zero-digit>
UMI Grouping
Overview
fgumi group assigns reads that appear to come from the same original molecule to the same group by writing a shared Molecular Identifier (MI) tag. Grouping relies on template-coordinate sort order.
This page describes:
- How reads and templates are filtered before grouping
- How mapping coordinates and UMIs identify reads from the same molecule
- Template-coordinate sort order
- Cell barcode support
- Metrics output
Filtering Reads and Templates
A read is a single sequenced strand. A template is all reads sharing the same query name (typically a read pair).
| Concept | Definition | Example |
|---|---|---|
| Read | A single sequenced strand (R1 or R2) | @read123/1 |
| Template | The full fragment, represented by both reads in a pair | @read123 includes both /1 and /2 |
Reads and templates are filtered before grouping to prevent splitting reads from a single molecule into separate groups.
Individual reads are filtered if:
- Flagged as secondary (unless
--include-secondary) - Flagged as supplementary (unless
--include-supplementary)
All reads for a template are filtered if:
- All reads for the template are unmapped (unless
--allow-unmapped) - Any non-secondary, non-supplementary read has mapping quality <
--min-map-q - Any UMI sequence contains one or more
Nbases --min-umi-lengthis specified and the UMI does not meet the length requirement
Grouping Unmapped Reads
By default, templates where all reads are unmapped are excluded from grouping. Pass --allow-unmapped
to include them. This is useful for workflows where some templates genuinely fail to align
(e.g. cell-free DNA fragments that fall outside the target region) but should still be counted
and may share UMIs with mapped templates from the same molecule:
fgumi group \
--input sorted.bam \
--output grouped.bam \
--strategy adjacency \
--allow-unmapped
Grouping Strategies
Grouping is performed by one of four strategies:
identity
Only reads with identical UMI sequences are grouped together. This is simpler and faster than other strategies, but should usually be avoided because sequencing errors in the UMI will split reads from the same molecule into separate groups. Useful for data exploration.
edit
Reads are clustered into groups such that each read within a group has at least one other read in the group with <= --edits differences, and there are no inter-group pairings with <= --edits differences. Effective when there are small numbers of reads per UMI, but breaks down at very high UMI coverage.
adjacency
A version of the directed adjacency method described in umi_tools that allows for errors between UMIs but only when there is a count gradient. Recommended for most simplex and CODEC workflows.
paired
Similar to adjacency but for duplex sequencing where each template has two UMIs (one from each strand). Expects UMI sequences stored in a single tag separated by a hyphen (e.g. ACGT-CCGG). Allows one UMI to be absent (e.g. ACGT- or -ACGT).
The molecular IDs produced have structure: {base}/{A|B}. For example, UMI pairs AAAA-GGGG and GGGG-AAAA map to 1/A and 1/B respectively. See Tracking Reads for details. Recommended for duplex workflows.
The edit, adjacency, and paired strategies use the --edits parameter to control matching of non-identical UMIs.
Cell Barcode Support
When processing data with cell barcodes (e.g. single-cell sequencing), reads at the same genomic position are partitioned by cell barcode before UMI assignment. This ensures that reads from different cells are never grouped together, even if they share a UMI and mapping position.
The cell barcode is read from the standard CB tag. No correction or
error-handling is performed on cell barcodes — they must be corrected upstream before grouping.
Cell barcodes are detected automatically across the entire pipeline — no additional flags are needed. The consensus callers validate that all source reads in a group share the same cell barcode and propagate it to the output consensus read.
Metrics Output
fgumi group can emit three types of metrics files. They can be specified individually or all at
once with the --metrics prefix flag.
Using --metrics (recommended)
The -M/--metrics flag writes all three metrics files under a single prefix in one step:
fgumi group \
--input sorted.bam \
--output grouped.bam \
--strategy adjacency \
--metrics my_sample
This produces:
my_sample.family_sizes.txt— histogram of UMI family sizesmy_sample.grouping_metrics.txt— overall grouping statisticsmy_sample.position_group_sizes.txt— histogram of UMI families per genomic position
Using individual flags
The three metrics can also be written to explicit paths:
fgumi group \
--input sorted.bam \
--output grouped.bam \
--strategy adjacency \
--family-size-histogram family_sizes.txt \
--grouping-metrics grouping_metrics.txt
Note: position_group_sizes.txt is only available via --metrics. The individual flags
--family-size-histogram and --grouping-metrics can be used alongside --metrics.
Family sizes
The family_sizes.txt file is a histogram of how many reads belong to each UMI family. A large
fraction of singleton families may indicate UMI collisions, over-sequencing, or UMI extraction
errors.
Grouping metrics
The grouping_metrics.txt file contains summary statistics about the grouping run, including
total reads, accepted reads, discarded reads by reason, and UMI assignment counts.
Position group sizes
The position_group_sizes.txt file is a histogram of how many distinct UMI families were
observed at each unique genomic position (coordinate + strand). A distribution skewed toward
large position groups may indicate high on-target duplication or UMI exhaustion.
Template-Coordinate Sort Order
fgumi group requires its input to be template-coordinate sorted. The header must advertise
SO:unsorted, GO:query, and SS:template-coordinate; without SS:template-coordinate the
input is treated as queryname-grouped (e.g. FASTQ-order output from fgumi extract) and
rejected with an actionable error pointing back here. fgumi group does not sort
internally — pre-sort with:
fgumi sort --order template-coordinate --input aligned.bam --output sorted.bam
The streaming grouper relies on records that share a position key being consecutive in the
input, which is what template-coordinate sort guarantees. Any other ordering (queryname,
coordinate, FASTQ-order) would split each true molecule across many small groups and assign
distinct MI values to reads that should share one.
For single-cell data, the CB cell barcode tag is automatically incorporated in the sort key,
keeping templates from different cells at the same locus separate:
fgumi sort --order template-coordinate --input aligned.bam --output sorted.bam
Template-coordinate order sorts reads by:
- The earlier unclipped 5’ coordinate of the read pair
- The higher unclipped 5’ coordinate of the read pair
- Strand orientation
- The cellular barcode (CB tag, if present)
- The molecular identifier (MI tag, if present)
- Read name
- Library (from read group)
- Whether R1 has the lower coordinates of the pair
Reads grouped by fgumi group with the same MI will share the same outer start/stop coordinates.
Because 5’ coordinates are strand-aware, reads from opposite strands with the same UMI and
position will not be grouped together (they belong to different strands of the same duplex
molecule).
See also: Consensus Calling, Duplex Consensus Calling, Best Practices
Tracking Reads through Grouping and Duplex Consensus Calling
This guide describes conventions for tracking reads from raw data through grouping and duplex consensus calling. It covers how molecular identifiers relate to strand assignment and how consensus tags encode single-strand and duplex information.
Top and Bottom Strand for Raw Reads
fgumi group assigns the same molecular ID to raw reads from the same source molecule, with trailing /A and /B to indicate which strand they belong to (top or bottom, AB or BA).
Convention: The /A raw reads are those where the 5’ unclipped position of read one (of the pair) is less than or equal to the 5’ unclipped position of read two. The 5’ unclipped position is relative to sequencing order, not the reference genome strand.
For example:
x: R1-----------------> <-------------------R2
y: R2-----------------> <-------------------R1
z: R1----------------->
<-----------------R2
xgets/A(R1’s 5’ end is at or before R2’s 5’ end)ygets/B(R1’s 5’ end is after R2’s 5’ end in sequencing order)zgets/A(even though fully overlapped, R1’s 5’ end is earlier)
Single-Strand Reads Relative to Duplex Consensus
fgumi duplex writes single-strand information into SAM tags for each duplex consensus read. Which single-strand consensus goes into the “AB” vs “BA” tags is determined as follows:
- Both strands present: Information for raw reads with
/Ain their molecular ID goes into “AB” tags;/Breads go into “BA” tags. - Only one strand present: The “AB” tags contain the single-strand consensus that was generated. The “BA” tags contain only per-read tags (no consensus data).
The duplex consensus sequence has the same strand orientation as the “AB” single-strand consensus.
Consensus Tags
SAM tags used for single-strand and duplex consensus reads:
| Value | AB Tag | BA Tag | Final Tag |
|---|---|---|---|
| Per-read depth | aD | bD | cD |
| Per-read min depth | aM | bM | cM |
| Per-read error rate | aE | bE | cE |
| Per-base depth | ad | bd | cd |
| Per-base error count | ae | be | ce |
| Per-base bases | ac | bc | (bases) |
| Per-base quals | aq | bq | (quals) |
Convention: The second letter in the tag is lowercase for per-base values and uppercase for per-read values.
Calling Consensus Reads
Overview
Reads with the same molecular identifier (MI tag) are examined base-by-base to determine the most likely base in the original source molecule. The consensus calling model has three steps:
- Adjusting input base qualities
- Computing the maximum posterior probability base
- Adjusting the output consensus base quality
Glossary
| Symbol | Description |
|---|---|
| Q | Phred-scaled base quality for a single base (measures sequencing error) |
| S_Q | Value subtracted from input base qualities (prior to capping) |
| M_Q | Maximum base quality cap (applied after shifting) |
| Err_pre | Phred-scaled error rate for errors before UMI integration (e.g. deamination, oxidation during library prep) |
| Err_post | Phred-scaled error rate for errors after UMI integration but before sequencing (e.g. amplification, target capture) |
| B_i | The base of the i-th read at a given position |
Step 1: Adjusting Input Base Qualities
Base qualities are assumed to represent the probability of a sequencing error. Two optional adjustments are applied:
- Shift: Subtract a fixed value from the phred-scaled qualities (e.g., Q30 with shift of 10 becomes Q20)
- Cap: Limit to a maximum phred-scaled value
Q' = min(Q - S_Q, M_Q)
These adjustments should only be used if input base qualities are systematically over-estimated.
The adjusted quality is converted to an error probability:
P_Q' = 10^(-Q'/10)
Then combined with the post-UMI error rate to produce a compound error probability covering all processes from UMI integration through sequencing:
P_Q'' = Err_post * (1 - P_Q') + (1 - Err_post) * P_Q' + (Err_post * P_Q' * 2/3)
This formula sums three terms:
- Error in post-UMI processes, no sequencing error
- No post-UMI error, but sequencing error
- Both errors occur, but the second doesn’t reverse the first (probability 2/3 for DNA with 4 bases)
Step 2: Computing the Consensus Base
For each position, the likelihood that the true base is A, C, G, or T is computed by multiplying across all reads:
L(Call=B) = ∏_i { P_Q''/3 if B ≠ B_i
{ 1 - P_Q'' if B = B_i
The likelihoods are normalized to posterior probabilities (assuming a uniform prior):
Post(Call=B) = L(Call=B) / Σ L(Call=C) for C in {A, C, G, T}
The base with the maximum posterior probability becomes the consensus call.
Step 3: Adjusting Output Quality
The consensus posterior is converted to an error probability and then modified to incorporate the pre-UMI error rate (errors before UMI integration, such as deamination or oxidation):
Pr_err = 1 - Post(Call)
Pr_err' = Err_pre * (1 - Pr_err) + (1 - Err_pre) * Pr_err + (Err_pre * Pr_err * 2/3)
Q_call = -10 * log10(Pr_err')
The final consensus base quality represents the probability of error across the entire process: from sample extraction through library preparation, UMI integration, amplification, and sequencing.
Any consensus base with quality below the minimum threshold is masked to N.
Caveats
- Each end of a pair is treated independently; overlapping bases within a pair are jointly called by default (disable with
--consensus-call-overlapping-bases false) - Indel errors in the reads are not considered in the consensus model
simplexandcodecdo not accept a--sort-orderflag; consensus reads are emitted as unmapped and should be sorted by the downstream pipeline (fgumi zipper+fgumi sort)
Duplex Consensus Calling
Overview
Duplex consensus calling takes reads generated from both strands of a double-stranded source molecule and produces consensus reads with extremely low error rates. This is the process used in duplex sequencing methods such as those described by Kennedy et al, where UMIs are attached to each end of the source molecule.
The mathematical model is similar to single-strand consensus calling, but the mechanics differ because reads from both strands must be combined.
Duplex consensus calling is run after grouping reads with fgumi group --strategy paired.
Process
Starting from a group of reads identified as originating from the same double-stranded molecule, the two strands are labeled A and B. The process proceeds through these steps:
- Split reads into four sub-groups: A1 (strand A, read 1), A2, B1, B2
- Unmap and revert to sequencing order
- Quality trim (optional, recommended)
- Mask remaining low-quality bases to
N - Trim to insert length to avoid reading into adapters
- Filter by CIGAR to ensure reads are in phase
- Call four single-strand consensus reads (one each for A1, A2, B1, B2)
- Call two duplex consensus reads by combining A1+B2 and A2+B1
Splitting Reads into Groups
Reads are split by strand of origin (A or B) and whether they are sequencing read 1 or 2. R1s from strand A correspond to R2s from strand B, and vice versa.
Quality Trimming
Reads can be end-trimmed to remove low-quality bases. This is highly recommended as it reduces disagreements in the consensus and fewer no-calls (Ns). Trimming uses the same running-sum algorithm as BWA.
Masking Low-Quality Bases
Bases below the minimum quality threshold are converted to Ns so they are not used in consensus calling. If quality trimming is disabled, reads are truncated to remove contiguous trailing Ns.
Trimming to Insert Length
Reads longer than the insert length read into adapter sequence. For duplex data, A1 and B2 reads may read into different adapter sequences. Calling consensus across different adapters produces many disagreements and no-calls, potentially causing consensus reads to be erroneously filtered. Reads are therefore trimmed to insert length before consensus calling.
CIGAR Filtering
Without multiple alignment, length errors (indels) in raw reads cause reads to be out of phase with each other. For example:
1: ACGTGACTGACTAGCTTTTTTT-AGACTAGCTACTACT
2: ACGTGACTGACTAGCTTTTTTT-AGACTAGCTACTACT
3: ACGTGACTGACTAGCTTTTTTTT-GACTAGCTACTACT
Read 3 has an extra T, causing many disagreements with reads 1 and 2.
To handle this, reads are grouped by compatible CIGAR alignments, and only the largest group is used for consensus. This is performed independently on A1+B2 and B1+A2 reads.
Calling Single-Strand Consensus Reads
Four single-strand consensus reads are generated (A1, A2, B1, B2) using the standard consensus calling model.
Calling Duplex Consensus Reads
The final duplex R1 and R2 are produced by merging the appropriate A and B reads base-by-base:
- Bases agree: quality = Q(A) + Q(B)
- Bases disagree, different qualities: base = higher quality base, quality = Q(higher) - Q(lower)
- Bases disagree, same quality: base is arbitrarily from A, quality = 2 (minimum Phred score)
The min-reads Parameter
For Simplex Consensus
fgumi simplex and fgumi filter accept a single --min-reads value.
For Duplex Consensus
fgumi duplex and fgumi filter accept one, two, or three --min-reads values. If fewer than three values are supplied, the last is repeated (e.g. 80 40 becomes 80 40 40, 10 becomes 10 10 10).
The values control:
- First value: minimum total raw reads across both single-strand consensuses for the final duplex read
- Second value: minimum reads for the single-strand consensus with more support
- Third value: minimum reads for the single-strand consensus with less support
If values two and three differ, the more stringent value must come first.
Example: --min-reads 7 3 1 requires:
- At least 7 total raw reads supporting the duplex consensus
- At least 3 raw reads for the better-supported single-strand consensus
- At least 1 raw read for the other single-strand consensus
Methylation Pipeline Guide
This guide describes how to process methylation sequencing data through fgumi’s consensus pipeline. It covers EM-Seq and TAPs/Illumina 5-base chemistries, for both simplex and duplex consensus calling workflows.
Background
Both EM-Seq and TAPs detect cytosine methylation by converting one class of cytosines to thymine, but they target opposite classes:
| EM-Seq | TAPs | |
|---|---|---|
| Chemistry | TET2 + APOBEC | TET oxidation + pyridine borane |
| What gets converted | Unmethylated C → T | Methylated C → T |
| C in read at ref-C | Methylated (protected) | Unmethylated (not a target) |
| T in read at ref-C | Unmethylated (converted) | Methylated (converted) |
Impact on UMI Processing
C→T conversion affects consensus calling: at a reference C position, reads showing T are not errors — they represent conversion events. Standard consensus calling would treat C/T disagreements as sequencing errors and penalize quality. Methylation mode recognizes these as conversion events and tracks per-base evidence through consensus calling.
UMI sequences:
- EM-Seq: UMIs should be synthesized with methylated cytosines (5mC) to protect them from enzymatic conversion. Unmethylated C in UMIs is a library prep issue.
- TAPs: UMIs are unaffected — synthetic oligonucleotides contain unmethylated cytosines, which TAPs does not convert.
Pipeline Overview
The methylation pipeline follows the same structure as the standard consensus pipeline, with additional flags at the consensus, re-alignment, and filter steps. Methylation mode is supported by simplex and duplex consensus callers. The codec caller does not support methylation mode.
Phase 1: FASTQ → Grouped BAM
extract → [correct] → fastq | aligner | zipper → sort → group
Phase 2: Grouped BAM → Filtered Consensus
simplex/duplex → fastq | aligner | zipper → filter → sort
Chemistry-Specific Steps
| Step | EM-Seq | TAPs |
|---|---|---|
| Alignment | bwameth (bisulfite-aware) | bwa mem (standard) |
| Consensus | --methylation-mode em-seq --ref | --methylation-mode taps --ref |
| Re-alignment zipper | --restore-unconverted-bases | (no additional flags) |
| Filter | --methylation-mode em-seq | --methylation-mode taps |
Workflow A: Random UMIs (No Fixed UMI Set)
This is the simpler case. Random UMIs (e.g., random 8-mers ligated during library prep) do not need correction against a whitelist.
Step 1: UMI Extraction
Extract UMIs from FASTQ. No methylation-specific flags needed here.
Simplex (single UMI per read pair):
fgumi extract \
--inputs r1.fq.gz r2.fq.gz \
--read-structures 8M+T +T \
--sample "sample_name" \
--library "library_name" \
--output unmapped.bam \
--threads 4
Duplex (UMI from both ends):
fgumi extract \
--inputs r1.fq.gz r2.fq.gz \
--read-structures 8M+T 8M+T \
--sample "sample_name" \
--library "library_name" \
--output unmapped.bam \
--threads 4
Step 2: Alignment
EM-Seq — use a bisulfite-aware aligner (bwameth) because unmethylated C→T conversion looks like bisulfite conversion:
fgumi fastq --input unmapped.bam --no-read-suffix \
| bwameth.py --reference ref.fa --threads 16 --interleaved /dev/stdin \
| samtools view -b \
| fgumi zipper --unmapped unmapped.bam --reference ref.fa --output aligned.bam
TAPs — use a standard aligner (bwa mem) because only methylated Cs are converted, leaving most Cs intact:
fgumi fastq --input unmapped.bam \
| bwa mem -t 16 -p -K 150000000 -Y ref.fa - \
| fgumi zipper --unmapped unmapped.bam --reference ref.fa --output aligned.bam
Step 3: Sort
fgumi sort \
--input aligned.bam \
--output sorted.bam \
--order template-coordinate \
--threads 8 \
--max-memory 4G
Step 4: UMI Grouping
Simplex:
fgumi group \
--input sorted.bam \
--output grouped.bam \
--strategy adjacency \
--edits 1 \
--family-size-histogram fam_sizes.txt \
--threads 8
Duplex:
fgumi group \
--input sorted.bam \
--output grouped.bam \
--strategy paired \
--edits 1 \
--family-size-histogram fam_sizes.txt \
--threads 8
Step 5: Consensus Calling
Use --methylation-mode and --ref to enable methylation-aware consensus.
Simplex:
fgumi simplex \
--input grouped.bam \
--output consensus.bam \
--min-reads 1 \
--min-input-base-quality 20 \
--output-per-base-tags \
--methylation-mode <em-seq|taps> \
--ref ref.fa \
--threads 8
Duplex:
fgumi duplex \
--input grouped.bam \
--output consensus.bam \
--min-reads 1 \
--min-input-base-quality 20 \
--output-per-base-tags \
--methylation-mode <em-seq|taps> \
--ref ref.fa \
--threads 8
Step 6: Re-alignment
Consensus reads are unmapped and must be re-aligned.
EM-Seq — use --restore-unconverted-bases so that bases normalized during consensus (T→C at ref-C positions) are restored before bisulfite-aware re-alignment:
fgumi fastq --input consensus.bam --no-read-suffix \
| bwameth.py --reference ref.fa --threads 16 --interleaved /dev/stdin \
| samtools view -b \
| fgumi zipper --unmapped consensus.bam --reference ref.fa --restore-unconverted-bases --output consensus.mapped.bam
TAPs:
fgumi fastq --input consensus.bam \
| bwa mem -t 16 -p -K 150000000 -Y ref.fa - \
| fgumi zipper --unmapped consensus.bam --reference ref.fa --output consensus.mapped.bam
Step 7: Filtering
Simplex filtering:
fgumi filter \
--input consensus.mapped.bam \
--output filtered.bam \
--ref ref.fa \
--min-reads 3 \
--max-base-error-rate 0.1 \
--max-no-call-fraction 0.2 \
--min-methylation-depth 3 \
--methylation-mode <em-seq|taps> \
--min-conversion-fraction 0.9 \
--reverse-per-base-tags \
--threads 8
Duplex filtering:
fgumi filter \
--input consensus.mapped.bam \
--output filtered.bam \
--ref ref.fa \
--min-reads 10,5,3 \
--max-base-error-rate 0.1 \
--max-no-call-fraction 0.2 \
--min-methylation-depth 10,5,3 \
--require-single-strand-agreement \
--require-strand-methylation-agreement \
--methylation-mode <em-seq|taps> \
--min-conversion-fraction 0.9 \
--reverse-per-base-tags \
--threads 8
Step 8: Final Sort
fgumi sort \
--input filtered.bam \
--output final.bam \
--order coordinate \
--threads 8
Workflow B: Fixed UMIs (Known UMI Set)
When UMIs come from a fixed set (e.g., a synthesized pool of known sequences), add a correction step before alignment. This maps observed UMIs back to the correct whitelist entry.
Step 1: UMI Extraction
Same as Workflow A.
Step 2: UMI Correction
Correct UMIs against the known whitelist:
fgumi correct \
--input unmapped.bam \
--output corrected.bam \
--umi-files known_umis.txt \
--max-mismatches 1 \
--min-distance 1 \
--metrics correction_metrics.txt \
--threads 8
If your UMI design includes unmethylated cytosines, add --allow-c-to-t. This flag applies uniformly across all UMI segments regardless of read-pair index, since both R1 and R2 UMI segments are in forward orientation. Only C-to-T tolerance is needed; G-to-A tolerance is not required.
Steps 3-8: Alignment through Final Sort
After correction, the remaining steps are the same as Workflow A (steps 2-8).
Output Tags
When methylation mode is enabled, consensus reads carry additional BAM tags for methylation evidence.
Simplex Output Tags
| Tag | Type | Description |
|---|---|---|
MM | Z | SAM-spec methylation modification calls (sparse format) |
ML | B:C | Methylation modification probabilities (companion to MM) |
cu | B:s | Per-base unconverted count (reads showing C at ref-C) |
ct | B:s | Per-base converted count (reads showing T at ref-C) |
Duplex Output Tags
All simplex tags above (combined from both strands), plus per-strand tags:
| Tag | Type | Description |
|---|---|---|
am | Z | AB strand methylation calls (MM format, no ML companion) |
bm | Z | BA strand methylation calls (MM format, no ML companion) |
au | B:s | AB strand unconverted count |
at | B:s | AB strand converted count |
bu | B:s | BA strand unconverted count |
bt | B:s | BA strand converted count |
MM/ML Probability Interpretation
The cu and ct count tags have the same meaning in both chemistries:
cu: reads showing C (unconverted) at a reference C positionct: reads showing T (converted) at a reference C position
The MM/ML probability differs:
- EM-Seq:
prob = cu / (cu + ct)— higher probability = more methylated (C stayed as C because it was protected) - TAPs:
prob = ct / (cu + ct)— higher probability = more methylated (C was converted to T because it was methylated)
The MM/ML tags follow the SAM-spec methylation format and are compatible with downstream methylation analysis tools.
Filter Options
The filter command provides methylation-specific options. These operate on the cu/ct/au/at/bu/bt count tags emitted by methylation-aware consensus calling.
--min-methylation-depth
Per-base masking based on methylation evidence depth. Bases where cu[i] + ct[i] is below the threshold are masked to N.
Accepts 1-3 comma-delimited values for duplex reads, following the same convention as --min-reads:
| Values | Meaning |
|---|---|
5 | 5 for all levels |
10,5 | 10 for duplex combined, 5 for each strand |
10,5,3 | 10 for duplex combined, 5 for AB strand, 3 for BA strand |
For simplex reads, only the first value is used.
--require-strand-methylation-agreement
Duplex-only, per-base masking. Requires --ref.
At each CpG dinucleotide in the reference, compares the methylation call from the top strand (AB: au/at at the C position) with the call from the bottom strand (BA: bu/bt at the G position). If one strand calls methylated and the other calls unmethylated, both positions of the CpG are masked to N.
This is analogous to --require-single-strand-agreement but specific to methylation status at CpG sites rather than raw base identity.
--min-conversion-fraction
Read-level filter. Requires --ref and --methylation-mode. Accepts a value between 0.0 and 1.0.
Computes the conversion fraction at non-CpG reference cytosine positions across the read:
- EM-Seq (
--methylation-mode em-seq): checksct / (cu + ct) >= threshold. Non-CpG cytosines are expected to be unmethylated and therefore converted. High conversion = good enzymatic conversion efficiency. - TAPs (
--methylation-mode taps): checkscu / (cu + ct) >= threshold. Non-CpG cytosines are expected to be unmethylated and therefore not converted. High non-conversion at non-CpG = good TAPs specificity.
CpG positions are excluded from both calculations because they may have variable methylation status.
Recommended Parameters
Simplex (Moderate Stringency)
fgumi simplex --min-reads 1 --min-input-base-quality 20 --output-per-base-tags \
--methylation-mode <em-seq|taps> --ref ref.fa
fgumi filter --ref ref.fa --min-reads 3 --max-base-error-rate 0.1 --min-methylation-depth 3 \
--methylation-mode <em-seq|taps> --min-conversion-fraction 0.9
Duplex (High Specificity)
fgumi duplex --min-reads 1 --min-input-base-quality 20 --output-per-base-tags \
--methylation-mode <em-seq|taps> --ref ref.fa
fgumi filter --ref ref.fa --min-reads 10,5,3 --max-base-error-rate 0.1 --min-methylation-depth 10,5,3 \
--require-single-strand-agreement --require-strand-methylation-agreement \
--methylation-mode <em-seq|taps> --min-conversion-fraction 0.9
Deduplication (No Consensus)
For workflows that mark duplicates without consensus calling:
fgumi dedup \
--input sorted.bam \
--output deduped.bam \
--metrics metrics.txt
Troubleshooting
Low Family Sizes / Too Many UMI Groups
If family size histograms show many singletons:
- Check that
--editsis appropriate for your UMI length - For fixed UMIs, review correction metrics to see how many UMIs are being corrected vs rejected
- EM-Seq only: verify that UMI sequences are synthesized with methylated cytosines to protect them from enzymatic conversion
Missing MM/ML Tags on Output
Ensure both --methylation-mode and --ref are provided to the consensus caller. The reference FASTA must have an accompanying .dict file (generate with samtools dict if missing).
Unexpected Masking from Strand Methylation Agreement
--require-strand-methylation-agreement only applies to duplex reads at CpG sites. If you see excessive masking:
- Check that your library has adequate duplex coverage at CpG sites
- Consider whether strand-specific methylation differences are biologically expected (e.g., imprinted regions)
- This filter requires both strands to have evidence — positions with zero evidence on either strand are not masked
Reads Filtered by Conversion Fraction
If many reads fail --min-conversion-fraction:
- EM-Seq: this indicates potential issues with enzymatic conversion efficiency
- TAPs: this indicates non-CpG cytosines are being converted, suggesting insufficient TAPs specificity
- Try lowering the threshold (e.g., 0.8 instead of 0.9)
- Check the overall conversion rate in your library QC metrics
- Reads with no non-CpG cytosine positions (e.g., very short reads aligned to AT-rich regions) automatically pass this filter
Using the Wrong Methylation Mode
If you use --methylation-mode em-seq for TAPs data (or vice versa), the methylation probabilities will be inverted — methylated positions will show low probability and vice versa. If downstream analysis shows unexpected methylation patterns, verify you used the correct mode for your chemistry.
fgumi Best Practice FASTQ -> Consensus Pipeline
This document describes the recommended best practice pipeline for processing FASTQ files through to consensus sequences using fgumi.
Tools Required
This pipeline uses only fgumi and a read aligner:
- fgumi (version 0.1 or higher)
- bwa mem (version 0.7.17 or higher recommended)
Unlike fgbio-based pipelines, no samtools is required - fgumi provides native fastq, sort, and merge commands.
Common Configuration Options
Compression Level
fgumi supports compression levels 1-12 for BAM output:
| Use Case | Level | Notes |
|---|---|---|
| Final outputs | 6-9 | Balance of size and speed |
| Intermediate files | 1 | Fast compression, larger files |
| Piped commands | 1 | Minimize CPU overhead |
Set with --compression-level N on any command that writes BAM.
Threading
All major fgumi commands support multi-threading via --threads N:
# Single-threaded (default, optimized fast path)
fgumi group --input in.bam --output out.bam --strategy adjacency
# Multi-threaded with 8 threads
fgumi group --input in.bam --output out.bam --strategy adjacency --threads 8
Thread allocation is automatically optimized per-command based on workload profiling.
Memory
fgumi’s memory model differs significantly from fgbio’s JVM -Xmx. In particular, --queue-memory is per-thread by default and controls only pipeline queue backpressure — actual process memory will be higher. See the Performance Tuning Guide for detailed guidance, including a comparison table for fgbio users.
Boolean Flags
All boolean flags accept the following values (case-insensitive): true/false, yes/no,
y/n, t/f. For example:
fgumi filter --require-single-strand-agreement yes ...
fgumi simplex --output-per-base-tags true ...
fgumi group --allow-unmapped y ...
Pipeline Overview
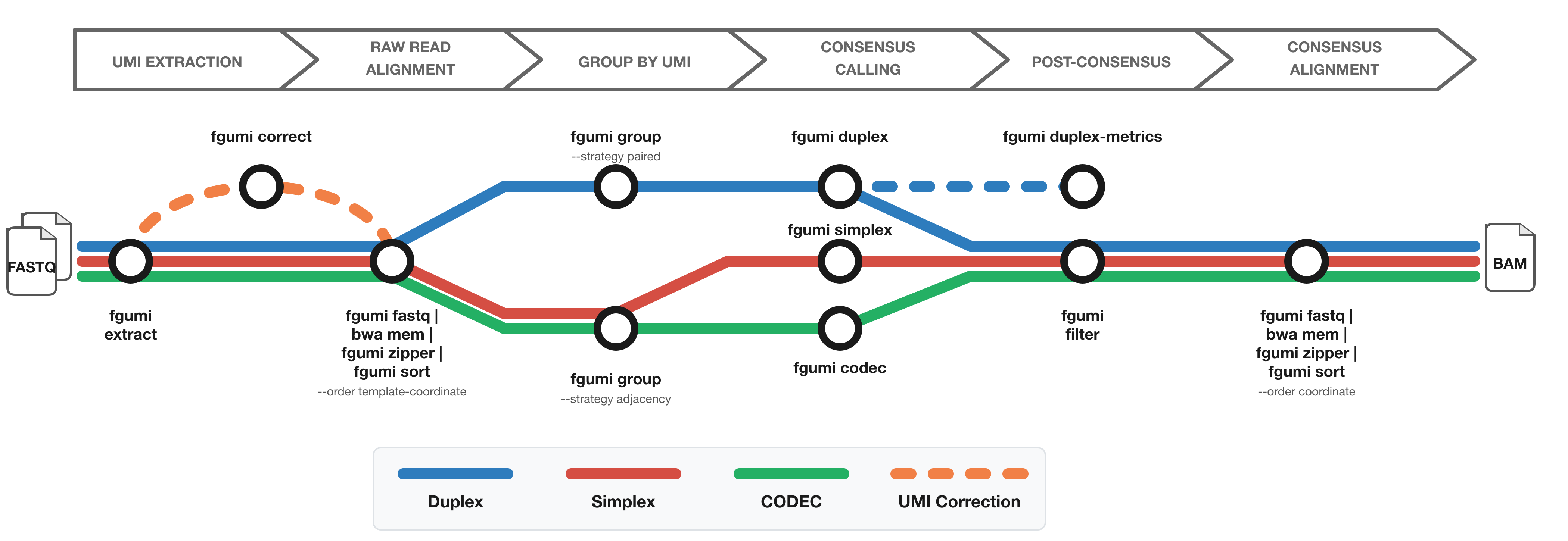
The diagram shows the workflow from FASTQ files to filtered consensus reads:
- Red: Simplex (single-strand) consensus
- Blue: Duplex (double-strand) consensus
- Green: CODEC consensus
- Orange: Optional UMI correction for fixed UMI sets
Phase 1: FASTQ → Grouped BAM
graph TD;
A["fgumi extract"]-->B["fgumi fastq | bwa mem | fgumi zipper"];
B-->C["fgumi sort"];
C-->D["fgumi merge (optional)"];
D-->E["fgumi group"];
Phase 2a: Grouped BAM → Filtered Consensus (R&D Version)
graph TD;
A["fgumi simplex/duplex"]-->B["fgumi fastq | bwa mem | fgumi zipper"];
B-->C["fgumi filter | fgumi sort"];
Phase 2b: Aligned BAM → Filtered Consensus (High-Throughput Version)
graph TD;
A["fgumi simplex/duplex"]-->B["fgumi fastq | bwa mem | fgumi zipper | fgumi filter | fgumi sort"];
Phase 1: FASTQ to Grouped BAM
Step 1.1: UMI Extraction
Convert FASTQ files to unmapped BAM with UMI extraction:
fgumi extract \
--inputs r1.fq.gz r2.fq.gz \
--read-structures 8M+T +T \
--sample "sample_name" \
--library "library_name" \
--output unmapped.bam \
--threads 4
Key parameters:
--read-structures: Define UMI and template positions (e.g.,8M+T= 8bp UMI + template)
For dual-index UMIs (duplex sequencing), use paired read structures:
fgumi extract \
--inputs r1.fq.gz r2.fq.gz \
--read-structures 8M+T 8M+T \
--sample "sample_name" \
--library "library_name" \
--output unmapped.bam
Optional: UMI Error Correction
For fixed/known UMI sets, correct sequencing errors before alignment:
fgumi correct \
--input unmapped.bam \
--output corrected.bam \
--umi-files known_umis.txt \
--min-distance 1
Step 1.2: Alignment
Align reads using the fgumi fastq + zipper pipeline:
fgumi fastq --input unmapped.bam \
| bwa mem -t 16 -p -K 150000000 -Y ref.fa - \
| fgumi zipper --unmapped unmapped.bam --reference ref.fa --output aligned.bam
Key points:
fgumi fastqconverts BAM to interleaved FASTQ for the aligner-ptells bwa mem to expect interleaved paired-end reads-K 150000000sets batch size (improves reproducibility)-Yis critical: Use soft-clipping for supplementary alignments to preserve basesfgumi zippertransfers tags from unmapped BAM to aligned readsfgumi zipperaccepts SAM or BAM on stdin or--input. For best performance, pipe uncompressed BAM from the aligner (e.g.bwa-mem3 mem --bam=0); SAM is fine for aligners that can’t emit BAM
For large files, add threading:
fgumi fastq --input unmapped.bam --threads 4 \
| bwa mem -t 16 -p -K 150000000 -Y ref.fa - \
| fgumi zipper --unmapped unmapped.bam --reference ref.fa --output aligned.bam --threads 4
Step 1.3: Sorting
Sort into template-coordinate order before grouping:
fgumi sort \
--input aligned.bam \
--output sorted.bam \
--order template-coordinate \
--threads 8 \
--max-memory 4G
For single-cell data, the CB cell barcode tag is automatically included in the
template-coordinate sort key, keeping templates from different cells at the same locus separate:
fgumi sort \
--input aligned.bam \
--output sorted.bam \
--order template-coordinate \
--threads 8
Step 1.3b: (Optional) Merging Multiple BAMs
When processing multiple lanes or flowcells separately, merge the sorted BAMs before grouping.
fgumi merge performs an efficient k-way merge without re-sorting:
fgumi merge \
--order template-coordinate \
--output merged.bam \
lane1_sorted.bam lane2_sorted.bam lane3_sorted.bam
For large numbers of files, use --input-list:
fgumi merge \
--order template-coordinate \
--input-list bam_paths.txt \
--output merged.bam
For single-cell data, the CB cell barcode tag is automatically included in the merge key.
All inputs must be sorted in the same order as --order. Do not use samtools merge for
template-coordinate BAMs — it does not understand the tc tag that fgumi zipper adds, and
will produce incorrect ordering.
Step 1.4: UMI Grouping
Group reads by UMI using the appropriate strategy:
For simplex/single-UMI workflows:
fgumi group \
--input sorted.bam \
--output grouped.bam \
--strategy adjacency \
--edits 1 \
--metrics group_metrics \
--threads 8
For duplex/paired-UMI workflows:
fgumi group \
--input sorted.bam \
--output grouped.bam \
--strategy paired \
--edits 1 \
--metrics group_metrics \
--threads 8
The --metrics PREFIX flag writes all three metrics files in one step:
PREFIX.family_sizes.txt— family size histogramPREFIX.grouping_metrics.txt— grouping statisticsPREFIX.position_group_sizes.txt— UMI families per genomic position
These can also be written to explicit paths with --family-size-histogram and
--grouping-metrics.
For workflows with unmapped templates (e.g., some cfDNA assays):
fgumi group \
--input sorted.bam \
--output grouped.bam \
--strategy adjacency \
--allow-unmapped \
--metrics group_metrics
By default, templates where all reads are unmapped are excluded. --allow-unmapped includes
them so their UMIs are still tracked and grouped with any mapped reads from the same molecule.
Step 1.5: (Optional) QC Metrics Before Consensus
For simplex libraries, collect QC metrics from the grouped BAM:
fgumi simplex-metrics \
--input grouped.bam \
--output simplex_metrics \
--min-reads 3
This produces simplex_metrics.family_sizes.txt, simplex_metrics.simplex_yield_metrics.txt,
simplex_metrics.umi_counts.txt, and optionally a PDF plot. The yield metrics show how the
number of callable consensus reads scales with sequencing depth (computed at 5%, 10%, …, 100%
of reads), so you can assess whether deeper sequencing would materially improve yield.
For duplex libraries, use duplex-metrics:
fgumi duplex-metrics \
--input grouped.bam \
--output duplex_metrics
Phase 2a: R&D Pipeline (Separate Consensus and Filtering)
This approach generates an intermediate consensus BAM, allowing you to experiment with different filtering parameters without re-running consensus calling.
Step 2a.1: Consensus Calling
Simplex consensus:
fgumi simplex \
--input grouped.bam \
--output consensus.bam \
--min-reads 1 \
--min-input-base-quality 20 \
--output-per-base-tags true \
--threads 8
Duplex consensus:
fgumi duplex \
--input grouped.bam \
--output consensus.bam \
--min-reads 1 \
--min-input-base-quality 20 \
--output-per-base-tags true \
--threads 8
Key parameters:
--min-reads 1: Keep all consensus reads (filter later)--output-per-base-tags true: Enable per-base filtering downstream--min-input-base-quality: Minimum quality for input bases (default: 10)
Note: --output-per-base-tags accepts true/false, yes/no, y/n, or t/f.
Step 2a.2: Re-alignment
Consensus reads are unmapped and must be re-aligned:
fgumi fastq --input consensus.bam \
| bwa mem -t 16 -p -K 150000000 -Y ref.fa - \
| fgumi zipper --unmapped consensus.bam --reference ref.fa --output consensus.mapped.bam
Step 2a.3: Filtering
Filter consensus reads with desired stringency:
Simplex filtering:
fgumi filter \
--input consensus.mapped.bam \
--output filtered.bam \
--ref ref.fa \
--min-reads 3 \
--max-read-error-rate 0.025 \
--max-base-error-rate 0.1 \
--min-base-quality 40 \
--max-no-call-fraction 0.2 \
--reverse-per-base-tags \
--threads 8
Duplex filtering (with strand-specific thresholds):
fgumi filter \
--input consensus.mapped.bam \
--output filtered.bam \
--ref ref.fa \
--min-reads 10,5,3 \
--max-read-error-rate 0.025 \
--max-base-error-rate 0.1 \
--min-base-quality 40 \
--max-no-call-fraction 0.2 \
--reverse-per-base-tags \
--require-single-strand-agreement true \
--threads 8
For duplex, --min-reads 10,5,3 means:
- 10 raw reads minimum for final duplex consensus
- 5 raw reads minimum for AB single-strand consensus
- 3 raw reads minimum for BA single-strand consensus
Step 2a.4: Final Sort (if needed)
Sort to coordinate order for downstream tools:
fgumi sort \
--input filtered.bam \
--output final.bam \
--order coordinate \
--threads 8
Phase 2b: Aligned BAM → Filtered Consensus (High-Throughput Version)
For production use where filtering parameters are established, combine steps for better throughput.
Stage 1: Group and call consensus in a single pipe:
fgumi group --input aligned.bam --strategy adjacency --threads 4 --compression-level 1 \
| fgumi simplex --input /dev/stdin --min-reads 1 --output-per-base-tags true \
--output consensus.bam --threads 4 --compression-level 1
Stage 2: Align, filter, and sort in a single pipe:
fgumi fastq --input consensus.bam \
| bwa mem -t 16 -p -K 150000000 -Y ref.fa - \
| fgumi zipper --unmapped consensus.bam --reference ref.fa \
| fgumi filter --input /dev/stdin --ref ref.fa --min-reads 3 \
| fgumi sort --input /dev/stdin --output filtered.bam --order coordinate --threads 4
Note: The two stages cannot be combined into a single pipeline because fgumi zipper --unmapped needs random access to the consensus BAM. For most use cases, the R&D pipeline with intermediate files provides better debuggability and flexibility.
Alternative: Deduplication Without Consensus
For workflows that need UMI-aware duplicate marking without consensus calling (e.g., when downstream tools handle deduplication differently, or for QC purposes), use fgumi dedup:
graph TD;
A["fgumi extract"]-->B["fgumi fastq | bwa mem | fgumi zipper"];
B-->C["fgumi sort --order template-coordinate"];
C-->D["fgumi dedup"];
Dedup Pipeline
# Step 1: Extract UMIs from FASTQ
fgumi extract \
--inputs r1.fq.gz r2.fq.gz \
--read-structures 8M+T 8M+T \
--sample "sample_name" \
--library "library_name" \
--output unmapped.bam
# Step 2: Align reads (fgumi zipper adds required `tc` tag)
fgumi fastq --input unmapped.bam \
| bwa mem -t 16 -p -K 150000000 -Y ref.fa - \
| fgumi zipper --unmapped unmapped.bam --reference ref.fa --output aligned.bam
# Step 3: Sort with fgumi (required - samtools sort won't work)
fgumi sort --input aligned.bam --output sorted.bam --order template-coordinate
# Step 4: Mark duplicates
fgumi dedup --input sorted.bam --output deduped.bam --metrics metrics.txt
Important: You MUST use fgumi zipper and fgumi sort before fgumi dedup:
fgumi zipperadds thetc(template-coordinate) tag to secondary/supplementary readsfgumi sort --order template-coordinatekeeps all alignments for a template together; downstreamfgumi dedupuses thetctag to validate inputsamtools sort --template-coordinatedoes NOT understand thetctag and will produce incorrect results for dedup
Dedup Options
# Remove duplicates instead of marking
fgumi dedup --input sorted.bam --output deduped.bam --remove-duplicates true
# Use a different UMI strategy (default: adjacency)
fgumi dedup --input sorted.bam --output deduped.bam --strategy paired --edits 1
# Write family size histogram
fgumi dedup --input sorted.bam --output deduped.bam \
--metrics metrics.txt \
--family-size-histogram histogram.txt
Recommended Parameters by Application
Variant Calling (High Sensitivity)
fgumi simplex --min-reads 1 --min-input-base-quality 10
fgumi filter --min-reads 2 --max-base-error-rate 0.2 --max-no-call-fraction 0.3
Variant Calling (High Specificity)
fgumi duplex --min-reads 1 --min-input-base-quality 20
fgumi filter --min-reads 10,5,3 --max-base-error-rate 0.1 --max-no-call-fraction 0.1 \
--require-single-strand-agreement true
Liquid Biopsy / ctDNA
fgumi duplex --min-reads 1 --min-input-base-quality 20
fgumi filter --min-reads 3,2,2 --max-base-error-rate 0.05 \
--require-single-strand-agreement true
Troubleshooting
Low Consensus Yield
- Check family size distribution with
--metricsonfgumi group - Lower
--min-readsthreshold - Verify UMI extraction with correct
--read-structures - Run
fgumi simplex-metricsorfgumi duplex-metricson the grouped BAM to assess yield curves
High Error Rates
- Increase
--min-input-base-qualityduring consensus calling - Tighten
--max-base-error-rateduring filtering - For duplex, use
--require-single-strand-agreement true
Memory Issues
- Use
--max-memorywithfgumi sortto limit RAM usage - Reduce
--threads(fewer threads = less memory) - Process in smaller batches
- See Performance Tuning for detailed guidance
See Also
- UMI Grouping — grouping strategies and cell barcode support
- Working with Metrics — metrics file formats and interpretation
- Performance Tuning — threading, memory, and compression
Performance Tuning Guide
fgumi provides three key options to optimize performance for your system: threading, memory management, and compression. This guide explains how to configure these options for different scenarios.
Coming from fgbio?
If you’re used to fgbio’s JVM-based memory model (java -Xmx4g), there are important differences in how fgumi manages memory:
| fgbio (JVM) | fgumi | |
|---|---|---|
| Memory control | -Xmx sets a hard ceiling on the entire process | --queue-memory controls pipeline queue backpressure |
| Enforcement | Hard limit — JVM throws OutOfMemoryError at the ceiling | Soft limit — triggers backpressure to slow producers |
| Scope | Total process memory (heap + off-heap) | Queue memory only; does not cover UMI data structures, decompressors, thread stacks, or working buffers |
| Scaling | Fixed regardless of threads | Per-thread by default (--queue-memory 768 --threads 8 = ~6 GB) |
| Recommendation | Set once and forget | Monitor RSS and adjust; use --queue-memory-per-thread false for a fixed total budget |
Key takeaway: fgumi’s actual process memory (RSS) will be higher than the --queue-memory value. When estimating memory needs, account for:
- Queue memory (controlled by
--queue-memory) - UMI grouping data structures (scales with UMI diversity and position depth)
- Per-thread decompressor and compressor instances
- Thread stacks and I/O buffers
For memory-constrained environments, start with --queue-memory-per-thread false and a conservative total budget, then increase if throughput is too low.
Threading Options
No-flag Fast Path (default)
- Usage: Omit
--threadsentirely - Behavior: Uses optimized single-threaded fast path with minimal overhead
- Best for: Small files, memory-constrained systems, debugging
Explicit Single-threaded Mode
- Usage:
--threads 1 - Behavior: Uses the unified pipeline with a single worker thread — same pipeline as
--threads Nbut with N=1; does not use the no-flag fast path - Best for: Isolating pipeline behavior in a single-threaded context
Multi-threaded Mode
- Usage:
--threads Nwhere N > 1 - Behavior: Uses unified 7-step pipeline with work-stealing scheduler
- Best for: Large files, high-performance systems, production workloads
Memory Management
fgumi’s unified memory management controls pipeline queue memory to prevent out-of-memory conditions while maintaining throughput.
Queue Memory Options
# Basic usage (768MB per thread - default)
fgumi filter --queue-memory 768 --queue-memory-per-thread true
# Human-readable formats
fgumi filter --queue-memory 2GB
fgumi filter --queue-memory 1024MiB
# Fixed total memory (no per-thread scaling)
fgumi filter --queue-memory 4096 --queue-memory-per-thread false
Memory Scaling Behavior
| Threads | Per-thread Mode | Fixed Mode |
|---|---|---|
| 1 | 768MB | 768MB |
| 4 | 3GB | 768MB |
| 8 | 6GB | 768MB |
| 16 | 12GB | 768MB |
Memory Validation
- System check: Warns if requesting >90% of available system memory
- Overflow protection: Prevents integer overflow with checked arithmetic
- Decimal support: Accepts formats like
1.5GBin addition to integers
Compression Options
Compression Level
- Range: 1 (fastest) to 12 (best compression)
- Default: 1 (fastest) for most commands;
fgumi mergedefaults to 6 - Usage:
--compression-level N
Compression Threading
- Default: Matches
--threadssetting - Override:
--compression-threads N - Best practice: Usually leave at default
I/O and Storage Tuning
For sequential workloads like BAM and FASTQ processing, I/O throughput is often the bottleneck — not CPU. Two areas to check: OS readahead and volume throughput.
OS Readahead
The Linux kernel prefetches file data into the page cache ahead of the application. The default readahead window is typically 128 KB, which fgumi’s decompression threads can easily outpace. When that happens the processing thread stalls waiting on disk.
Check the current readahead (in 512-byte sectors):
blockdev --getra /dev/nvme1n1 # e.g. 256 = 128 KB
For sequential BAM/FASTQ workloads, increasing to 4 MB eliminates most I/O stalls:
# 4 MB = 8192 sectors (requires root)
sudo blockdev --setra 8192 /dev/nvme1n1
This setting does not persist across reboots. Add it to a startup script or udev rule if needed.
--async-reader (Experimental)
When you cannot tune OS readahead — containers, managed cloud instances, network
mounts — --async-reader provides a similar benefit from userspace. It spawns a
dedicated I/O thread that reads raw bytes into a bounded queue ahead of the
decompression step, so processing threads do not block on disk.
fgumi group \
--async-reader \
--threads 8 \
--input reads.bam \
--output grouped.bam
--async-reader works with all input types: BAM files, BGZF/gzip/plain FASTQs,
and piped stdin. It is supported by all commands that read BAM/FASTQ input,
including sort. It is most effective when I/O latency is high (network storage,
cold page cache, small OS readahead). On systems where you can already set 4 MB+
readahead, the additional benefit is modest.
AWS EBS Volume Throughput
On AWS, gp3 volumes default to 125 MB/s throughput regardless of size. For BAM
processing this is often the binding constraint. Increasing to 300-500 MB/s is
inexpensive and has a large impact:
# Increase throughput on an existing volume (takes effect within minutes)
aws ec2 modify-volume \
--volume-id vol-0123456789abcdef0 \
--throughput 500
For sustained sequential I/O, also consider increasing IOPS (default 3000) if your
reads are small. Monitor with iostat -x 1 to confirm the volume is the bottleneck
before spending on higher provisioned throughput.
Scenario-Based Configurations
High-Throughput Server
Goal: Maximum processing speed for large datasets
fgumi filter \
--threads 16 \
--queue-memory 1GB \
--compression-level 3 \
--input large_dataset.bam \
--output filtered.bam
Rationale:
- High thread count for parallel processing
- Generous memory for pipeline buffers
- Lower compression for speed
Memory-Constrained Node
Goal: Minimize memory usage while maintaining reasonable performance
fgumi filter \
--threads 8 \
--queue-memory 512 \
--queue-memory-per-thread false \
--compression-level 6 \
--input dataset.bam \
--output filtered.bam
Rationale:
- Moderate thread count
- Fixed memory limit (512MB total)
- Default compression for balance
Fast Local SSD
Goal: Optimize for fast I/O with minimal compression overhead
fgumi filter \
--threads 8 \
--queue-memory 2GB \
--compression-level 1 \
--input dataset.bam \
--output filtered.bam
Rationale:
- High memory for large pipeline buffers
- Minimal compression (I/O not bottleneck)
Network Storage
Goal: Minimize network I/O with maximum compression
fgumi filter \
--async-reader \
--threads 4 \
--queue-memory 512 \
--compression-level 9 \
--input dataset.bam \
--output filtered.bam
Rationale:
--async-readerhides network I/O latency (see I/O and Storage Tuning)- Moderate threading to avoid overwhelming network
- Conservative memory usage
- Maximum compression to reduce network transfer
Development/Testing
Goal: Fast iteration with minimal resource usage
fgumi filter \
--queue-memory 256 \
--compression-level 1 \
--input small_test.bam \
--output test_output.bam
Rationale:
- Single-threaded for simplicity
- Minimal memory footprint
- Fast compression for quick turnaround
Verbose Logging
Use --verbose (or -v) to enable debug-level logging for any command:
fgumi group --verbose --input reads.bam --output grouped.bam
This is equivalent to setting RUST_LOG=debug. If RUST_LOG is explicitly set, it takes precedence over --verbose.
Advanced Pipeline Options
The following options are available on all multi-threaded pipeline commands. They are hidden from the default help text but can be useful for debugging and performance analysis.
Pipeline Statistics
fgumi group --pipeline-stats --input reads.bam --output grouped.bam
Prints detailed per-step timing, throughput, contention metrics, and per-thread work distribution at completion.
Scheduler Strategy
fgumi group --scheduler balanced-chase-drain --input reads.bam --output grouped.bam
Controls which scheduling strategy threads use for work assignment. The default (balanced-chase-drain) is recommended for most workloads. Available strategies:
| Strategy | Description |
|---|---|
balanced-chase-drain | Default. Balanced work distribution with output drain mode. |
fixed-priority | Static thread roles (reader, writer, workers). Simple baseline. |
chase-bottleneck | Threads dynamically follow work through the pipeline. |
Other experimental strategies are available (thompson-sampling, ucb, epsilon-greedy, etc.) but are not recommended for production use.
Deadlock Detection
# Adjust timeout (default: 10 seconds, 0 to disable)
fgumi group --deadlock-timeout 30 --input reads.bam --output grouped.bam
# Enable automatic recovery (default: detection only)
fgumi group --deadlock-recover --input reads.bam --output grouped.bam
The pipeline monitors for progress stalls. When no queue operations succeed for the timeout duration, diagnostic information is logged (queue depths, memory usage, per-queue timestamps).
With --deadlock-recover, the pipeline progressively doubles queue memory limits (2x, 4x, up to 8x) to resolve backpressure deadlocks, then restores original limits after 30 seconds of sustained progress.
Performance Monitoring
Memory Usage
- Monitor system memory usage during execution
- Watch for “exceeds available memory” warnings
- Adjust
--queue-memoryif seeing swap activity
Thread Utilization
- Use
htopor similar to monitor CPU usage - All threads should show activity during processing
- Consider reducing threads if not fully utilized
I/O Patterns
- Monitor disk I/O with
iotoporiostat -x 1 - If threads are idle waiting on I/O, increase OS readahead or try
--async-reader(see I/O and Storage Tuning) - Network storage may benefit from lower thread counts
- SSD storage can handle higher thread counts
Troubleshooting
Out of Memory Errors
- Reduce
--queue-memory - Set
--queue-memory-per-thread falsefor fixed limits - Reduce
--threads
Poor Performance
- Increase
--threadsif CPU usage is low - Increase
--queue-memoryif I/O bound - Reduce
--compression-levelif CPU bound - Check OS readahead and EBS throughput if disk I/O is the bottleneck (see I/O and Storage Tuning)
Pipeline Appears Stuck
If a command hangs without producing output:
- Check if a deadlock warning appears in the log (default timeout: 10 seconds)
- Run with
--verboseto see detailed pipeline activity - Run with
--pipeline-statsto see per-step metrics at completion - Try
--deadlock-recoverto allow automatic recovery from backpressure deadlocks - Reduce
--threads— fewer threads means simpler scheduling and less contention
System Memory Warnings
Requested memory 16GB exceeds 90% of system memory (14.4GB)
- Reduce memory allocation or add more RAM
- Consider using
--queue-memory-per-thread false
Command-Specific Considerations
Extract
- Benefits from high memory (large FASTQ processing)
- Compression level affects output size significantly
Zipper
- For best throughput, pipe uncompressed BAM from the aligner (e.g.
bwa-mem3 mem --bam=0). Uncompressed BAM skips SAM text formatting on the aligner side and SAM parsing on the zipper side, and adds only ~26 bytes of BGZF framing per ~64 KiB block - SAM input is fine for aligners that can’t emit BAM; compressed BAM on a pipe wastes CPU on both ends for data the sort step will re-compress anyway
- The zipper pipeline uses raw-byte merging internally: aligned records are not fully decoded and re-encoded unless the record actually needs modification, which eliminates a significant CPU bottleneck on high-throughput runs
Sort
- Uses an internal LoserTree (tournament tree) for k-way merging, which performs significantly better than a simple heap merge when the number of sorted runs is large
--max-memorycontrols how much RAM is used for sort buffers; increase for large files to reduce the number of intermediate merge passes- For template-coordinate sort with single-cell data, the
CBtag is included automatically --async-readeris supported and can improve Phase 1 (input reading) throughput when disk latency is high or the OS page cache readahead is small
Merge
fgumi mergeperforms a k-way merge using a LoserTree for efficient multi-file merging- Thread count (
--threads) controls compression parallelism, not merge concurrency - For template-coordinate merges with single-cell data, the
CBtag is included automatically
Group/Dedup
- Memory usage scales with UMI diversity and the number of reads at any given position
- Higher thread counts improve UMI processing
- The
--metrics PREFIXflag writes all grouping metrics in one step with minimal overhead
Simplex/Duplex Metrics
- Both
simplex-metricsandduplex-metricsare single-threaded; they do not benefit from--threads - Memory usage is proportional to the number of unique genomic positions in the input
Consensus (Simplex/Duplex/CODEC)
- Memory proportional to family sizes
- Benefits from balanced threading and memory
Filter
- Streaming operation benefits from pipeline memory
- Compression affects final output size
Migration from Legacy Parameters
If using deprecated --queue-memory-limit-mb:
# Old (deprecated)
fgumi group --queue-memory-limit-mb 4096
# New (recommended)
fgumi group --queue-memory 4096 --queue-memory-per-thread false
The new parameters provide better control and human-readable formats while maintaining backward compatibility.
Working with Metrics
fgumi commands produce structured metrics files for quality control and analysis. This guide covers the file formats, terminology, and how to work with the outputs.
Commands that Produce Metrics
| Command | Metrics Output | Flag |
|---|---|---|
filter | Filtering pass/fail statistics | --stats |
simplex | Consensus calling statistics | --stats |
duplex | Consensus calling statistics | --stats |
codec | Consensus calling statistics | --stats |
dedup | Deduplication metrics and family size histogram | --metrics, --family-size-histogram |
duplex-metrics | Comprehensive duplex QC metrics | --output (prefix) |
simplex-metrics | Comprehensive simplex QC metrics | --output (prefix) |
group | Family sizes, grouping metrics, position group sizes | --metrics (prefix), --family-size-histogram, --grouping-metrics |
See the Metrics Reference for field-level documentation of each metric type.
File Formats
Most metrics files are tab-separated values (TSV) with a header row. There are two formats:
Horizontal TSV (Most Commands)
A header row followed by a single data row. Used by dedup, codec, duplex-metrics,
simplex-metrics, and group.
total_templates unique_templates duplicate_templates duplicate_rate
25000 18750 6250 0.25
Vertical Key-Value (Simplex/Duplex)
The simplex and duplex commands use a three-column format with one metric per row:
key value description
raw_reads_considered 50000 Total raw reads considered from input file
raw_reads_used 41800 Total count of raw reads used in consensus reads
consensus_reads_emitted 12000 Total number of consensus reads (R1+R2=2) emitted
This format is compatible with fgbio’s CallMolecularConsensusReads output.
Filter Stats (Special Case)
The filter --stats output uses a two-column key-value format without a header row:
total_reads 10000
passed_reads 8542
pass_rate 0.8542
Group Metrics
fgumi group can produce three metrics files, all available together via --metrics PREFIX
(or individually with --family-size-histogram and --grouping-metrics):
| File | Description |
|---|---|
PREFIX.family_sizes.txt | Histogram of UMI family sizes (number of templates per family; on paired-end data a read pair counts as one template) |
PREFIX.grouping_metrics.txt | Overall grouping statistics: reads accepted/discarded and reasons |
PREFIX.position_group_sizes.txt | Histogram of how many UMI families were observed at each genomic position |
The position_group_sizes.txt file is only written when using --metrics; it is not available
through the individual --family-size-histogram/--grouping-metrics flags.
A large fraction of singleton families in family_sizes.txt may indicate UMI collisions,
over-sequencing, or incorrect read structures. A distribution skewed toward large values in
position_group_sizes.txt may indicate UMI exhaustion or very high on-target duplication.
Duplex Metrics
The duplex-metrics command uses specific terminology for family types:
| Prefix | Name | Definition |
|---|---|---|
| CS | Coordinate-Strand | Families defined by genome coordinates and strand only (no UMI information) |
| SS | Single-Stranded | Families defined by coordinates, strand, and UMI. Two SS families from the same molecule (e.g., 50/A and 50/B) are counted separately |
| DS | Double-Stranded | Collapsed across SS families from the same molecule. SS families from opposite strands become one DS family |
The duplex-metrics output files include:
| File | Description |
|---|---|
<prefix>.family_sizes.txt | Family size distribution by type (CS/SS/DS) |
<prefix>.duplex_family_sizes.txt | Duplex family sizes by A→B and B→A strand counts |
<prefix>.duplex_yield_metrics.txt | Summary QC metrics at subsampling levels (5%–100%) |
<prefix>.umi_counts.txt | UMI observation frequencies |
<prefix>.duplex_umi_counts.txt | Duplex UMI pair frequencies (optional, --duplex-umi-counts) |
<prefix>.duplex_qc.pdf | QC plots (requires R with ggplot2) |
Simplex Metrics
fgumi simplex-metrics collects comprehensive QC metrics for simplex (single-strand) sequencing
experiments. It takes a UMI-grouped BAM (output of fgumi group) as input:
fgumi simplex-metrics \
--input grouped.bam \
--output simplex_metrics \
--min-reads 3
Key options:
--min-reads N— minimum templates per SS family to count it as a consensus family in yield metrics (default: 1)--intervals FILE— restrict analysis to a BED or Picard interval list--description TEXT— sample name or description for PDF plot titles
The simplex-metrics output files include:
| File | Description |
|---|---|
<prefix>.family_sizes.txt | Family size distribution (CS and SS) |
<prefix>.simplex_yield_metrics.txt | Summary QC metrics at subsampling levels (5%–100%) |
<prefix>.umi_counts.txt | UMI observation frequencies |
<prefix>.simplex_qc.pdf | QC plots (requires R with ggplot2) |
Yield metrics are computed at multiple subsampling fractions (5%, 10%, …, 100%), allowing you to assess how yield scales with sequencing depth without re-running the full pipeline.
Reading Metrics Files
Python
import pandas as pd
# Read horizontal TSV (dedup, codec, duplex-metrics, group)
dedup_metrics = pd.read_csv("dedup_metrics.txt", sep="\t")
# Read vertical KV format (simplex, duplex)
consensus_stats = pd.read_csv("simplex_stats.txt", sep="\t")
# Access metrics by key:
# consensus_stats[consensus_stats["key"] == "consensus_reads_emitted"]["value"]
# Read filter stats (no header)
filter_stats = pd.read_csv("filter_stats.txt", sep="\t", header=None, names=["key", "value"])
R
# Read horizontal TSV
dedup_metrics <- read.table("dedup_metrics.txt", header=TRUE, sep="\t")
# Read vertical KV format
consensus_stats <- read.table("simplex_stats.txt", header=TRUE, sep="\t")
# Read filter stats (no header)
filter_stats <- read.table("filter_stats.txt", header=FALSE, sep="\t", col.names=c("key", "value"))
Comparing Metrics
Use fgumi compare metrics to compare metrics files between runs:
fgumi compare metrics file1.txt file2.txt --precision 6 --rel-tol 1e-6
This is useful for validating that pipeline changes produce equivalent results. See the compare documentation for details.
Note:
fgumi compareis a developer tool not included in standard builds. Build with--features compareto enable it:cargo build --release --features compare.
Migration from fgbio
fgumi is the Rust successor to fgbio for UMI-based tools. This guide maps fgbio tools to their fgumi equivalents and highlights key differences.
Command Mapping
| fgbio Tool | fgumi Command | Notes |
|---|---|---|
ExtractUmisFromBam | extract | Extracts directly from FASTQ (not BAM) |
CorrectUmis | correct | |
ZipperBams | zipper | Also replaces picard MergeBamAlignment; accepts SAM or BAM input |
SortBam | sort | Adds template-coordinate sort order with optional cell barcode key |
GroupReadsByUmi | group | Same strategies: identity, edit, adjacency, paired |
CallMolecularConsensusReads | simplex | |
CallDuplexConsensusReads | duplex | |
CallCodecConsensusReads | codec | |
FilterConsensusReads | filter | |
ClipBam | clip | |
CollectDuplexSeqMetrics | duplex-metrics | |
| (no equivalent) | simplex-metrics | New: simplex QC metrics (yield, family sizes, UMI counts) |
| (samtools merge) | merge | k-way merge of pre-sorted BAMs; supports all sort orders |
ReviewConsensusVariants | review |
Key Differences
Input Format
fgbio’s ExtractUmisFromBam takes an unmapped BAM as input. fgumi’s extract takes FASTQ files directly, which is more common in practice and avoids an unnecessary BAM conversion step.
Streaming Pipeline
fgumi supports Unix pipe-based streaming for the alignment workflow:
fgumi fastq --input unaligned.bam \
| bwa mem -p -K 150000000 -Y ref.fa - \
| fgumi zipper --unmapped unaligned.bam \
| fgumi sort --output sorted.bam --order template-coordinate
This replaces multiple separate fgbio/picard steps (SortBam, ZipperBams/MergeBamAlignment) with a single streaming pass. fgumi zipper accepts SAM or BAM on stdin or via --input; for best performance, pipe uncompressed BAM from the aligner (e.g. bwa-mem3 mem --bam=0).
Merging Multiple BAMs
fgbio users who relied on samtools merge to combine per-lane BAMs before grouping should use
fgumi merge instead. It performs an equivalent k-way merge and correctly handles
template-coordinate order with cell barcodes:
# fgbio/samtools workflow
samtools merge -n merged.bam lane1.bam lane2.bam lane3.bam
# fgumi equivalent (also supports template-coordinate and queryname sort orders)
fgumi merge --order template-coordinate --output merged.bam \
lane1.bam lane2.bam lane3.bam
If you produce a queryname-sorted output from fgumi merge (or from any
other source — fgumi extract, samtools sort -n, etc.), insert a
fgumi sort --order template-coordinate step before fgumi group,
fgumi dedup, or fgumi downsample. Unlike fgbio’s GroupReadsByUmi,
fgumi group does not sort internally — it requires its input to be
template-coordinate sorted with the SS:template-coordinate header tag,
and rejects any other sort order with an actionable error.
Simplex QC Metrics
fgbio has no equivalent to fgumi simplex-metrics. This command provides yield curves,
family size distributions, and UMI frequency statistics specifically for simplex sequencing
experiments, analogous to what duplex-metrics provides for duplex experiments.
Threading Model
fgumi uses a multi-threaded pipeline architecture where reading, processing, and writing happen
concurrently. Most commands accept --threads to control parallelism. See
Performance Tuning for details.
Grouping Strategies
fgumi supports the same four UMI assignment strategies as fgbio:
identity— exact UMI matching onlyedit— edit-distance clusteringadjacency— directional adjacency (recommended for most use cases)paired— paired adjacency for duplex workflows
The algorithms are equivalent but fgumi’s implementations are optimized for throughput.
Group Metrics
fgumi’s group command now produces a third metrics file beyond family sizes and grouping
metrics: position_group_sizes.txt, a histogram of how many UMI families appear at each
genomic position. This has no fgbio equivalent but is useful for detecting UMI exhaustion or
abnormal duplication patterns.
Use the --metrics PREFIX flag to write all three files in one step.
Metrics Compatibility
fgumi’s simplex and duplex stats output uses the same three-column key-value format as
fgbio’s CallMolecularConsensusReads, allowing direct comparison with fgumi compare metrics.
Sort Orders
fgumi’s sort command supports the same sort orders as fgbio:
coordinate— standard genomic coordinate sortqueryname— sort by read nametemplate-coordinate— sort by template 5’ positions (required input forgroup)
For single-cell data, fgumi sort --order template-coordinate automatically includes the CB
cell barcode tag in the sort key so that templates from different cells at the same locus are not
interleaved. fgbio’s template-coordinate sort does not support this.
Rejects BAM Sort Order
When --rejects is enabled on simplex, duplex, codec, or correct, fgumi writes
rejected records from worker threads in mutex-acquisition order, which is not guaranteed
to match input order under --threads > 1. Because of this, fgumi stamps the rejects BAM
header with SO:unsorted (and drops any GO/SS tags inherited from the input) so
downstream tools don’t assume the input’s sort order carried over.
fgbio’s equivalent tools copy the input header onto the rejects BAM unchanged, which can
leave a stale SO tag when more than one consensus-calling thread is used. If you were
relying on fgbio’s rejects header carrying the input’s sort order, sort the rejects BAM
explicitly after the fact.
Boolean Flag Values
fgumi boolean flags (e.g. --output-per-base-tags, --trim, --require-single-strand-agreement)
accept the following values: true/false, yes/no, y/n, t/f (case-insensitive).
fgbio uses standard true/false only.
Removed Options
The --sort-order flag has been removed from simplex and codec. Output sort order for
consensus reads is determined by the downstream pipeline step (zipper + sort), not by the
consensus caller itself.
What fgumi Does Not Replace
fgumi focuses on UMI-based tools. The following fgbio tools do not have fgumi equivalents:
- Non-UMI tools (e.g.,
TrimFastq,ErrorRateByReadPosition,EstimatePoolingFractions) - VCF tools (e.g.,
FilterSomaticVcf,HapTyper) - FASTQ/FASTA utilities (e.g.,
FastqToBam,HardMaskFasta)
Continue using fgbio for these tools.
Tool Reference
Auto-generated from fgumi command definitions.
ALIGNMENT
| Command | Description |
|---|---|
fastq | Convert BAM to FASTQ format |
zipper | Zip unmapped BAM with aligned BAM |
sort | Sort BAM file by coordinate, queryname, or template-coordinate |
merge | Merge pre-sorted BAM files into a single sorted BAM |
CONSENSUS
| Command | Description |
|---|---|
simplex | Call simplex consensus sequences from UMI-grouped reads |
duplex | Call duplex consensus sequences from UMI-grouped reads |
codec | Call CODEC consensus reads from grouped BAM |
DEDUP
| Command | Description |
|---|---|
dedup | Mark or remove PCR duplicates using UMI information |
GROUP
| Command | Description |
|---|---|
group | Group reads by UMI to identify reads from the same original molecule |
POST-CONSENSUS
| Command | Description |
|---|---|
filter | Filter consensus reads based on quality metrics |
clip | Clip overlapping reads in BAM files |
duplex-metrics | Collect QC metrics for duplex consensus reads |
review | Extract data to review variant calls from consensus reads |
simplex-metrics | Collect QC metrics for simplex sequencing data |
UMI EXTRACTION
| Command | Description |
|---|---|
extract | Extract UMIs from FASTQ and create unmapped BAM |
correct | Correct UMIs in a BAM file to a fixed set of UMIs |
UTILITIES
| Command | Description |
|---|---|
downsample | Downsample BAM by UMI family using streaming |
extract
Category: UMI EXTRACTION
Extract UMIs from FASTQ and create unmapped BAM
Description
Generates an unmapped BAM file from FASTQ files with UMI extraction.
Takes in one or more FASTQ files (optionally gzipped), each representing a different sequencing read (e.g. R1, R2, I1 or I2) and can use a set of read structures to allocate bases in those reads to template reads, sample indices, unique molecular indices, or to designate bases to be skipped over.
Only template bases will be retained as read bases (stored in the SEQ field) as specified by
the read structure.
Read Structures
Read structures are made up of <number><operator> pairs much like the CIGAR string in BAM files.
Five kinds of operators are recognized:
Tidentifies a template readBidentifies a sample barcode readMidentifies a unique molecular index readCidentifies a cell barcode readSidentifies a set of bases that should be skipped or ignored
The last <number><operator> pair may be specified using a + sign instead of number to denote
“all remaining bases”. This is useful if, e.g., FASTQs have been trimmed and contain reads of
varying length.
For example, to convert a paired-end run with an index read and where the first 5 bases of R1 are a UMI and the second five bases are monotemplate:
fgumi extract –input r1.fq r2.fq i1.fq –read-structures 5M5S+T +T +B
Alternatively, if reads are fixed length:
fgumi extract –input r1.fq r2.fq i1.fq –read-structures 5M5S65T 75T 8B
UMI Extraction
A read structure should be provided for each read of a template. For paired end reads, two read structures should be specified. The tags to store the molecular indices will be associated with the molecular index segment(s) in the read structure based on the order specified. If only one molecular index tag is given, then the molecular indices will be concatenated and stored in that tag. In the resulting BAM file each end of a pair will contain the same molecular index tags and values.
UMIs may be extracted from the read sequences, the read names, or both. If
--extract-umis-from-read-names is specified, any UMIs present in the read names are extracted;
read names are expected to be :-separated and the UMI is taken from the last field. At
least 8 fields must be present — the standard Illumina shape
@<instrument>:<run>:<flowcell>:<lane>:<tile>:<x>:<y>:<UMI>. Names with 9+ fields (e.g.
produced by demultiplexers that fold the sample index into the colon-separated portion) are
also handled, with the UMI still coming from the last field. Any + characters in the
extracted UMI are normalized to -. If UMI segments are present in the read structures those
will also be extracted. If UMIs are present in both, the final UMIs are constructed by first
taking the UMIs from the read names, then adding a hyphen, then the UMIs extracted from the
reads.
Arguments
| Flag | Description | Default |
|---|---|---|
-i, --inputs <INPUTS> | Input FASTQ files corresponding to each sequencing read (e.g. R1, I1, etc.) | required |
-o, --output <OUTPUT> | Output BAM file to be written | required |
-r, --read-structures <READ_STRUCTURES> | Read structures, one for each of the FASTQs (optional if 1-2 template-only FASTQs) | |
-q, --store-umi-quals <STORE_UMI_QUALS> | Store UMI base quality scores in the QX SAM tag | |
-C, --store-cell-quals <STORE_CELL_QUALS> | Store cell barcode base quality scores in the CY SAM tag | |
-Q, --store-sample-barcode-qualities <STORE_SAMPLE_BARCODE_QUALITIES> | Store the sample barcode qualities in the QT Tag | |
-n, --extract-umis-from-read-names <EXTRACT_UMIS_FROM_READ_NAMES> | Extract UMI(s) from read names and prepend to UMIs from reads | |
-a, --annotate-read-names <ANNOTATE_READ_NAMES> | Annotate read names with UMIs (appends “+UMIs” to read names) | |
-s, --single-tag <SINGLE_TAG> | Single tag to store all concatenated UMIs (in addition to per-segment tags) | |
--clipping-attribute <CLIPPING_ATTRIBUTE> | Tag containing adapter clipping position to adjust (e.g. ‘XT’ from MarkIlluminaAdapters) | |
--read-group-id <READ_GROUP_ID> | Read group ID to use in the file header | A |
--sample <SAMPLE> | The name of the sequenced sample | required |
--library <LIBRARY> | The name/ID of the sequenced library | required |
-b, --barcode <BARCODE> | Library or Sample barcode sequence | |
--platform <PLATFORM> | Sequencing Platform | illumina |
--platform-unit <PLATFORM_UNIT> | Platform unit (e.g. ‘flowcell-barcode.lane.sample-barcode’) | |
--platform-model <PLATFORM_MODEL> | Platform model to insert into the group header (ex. miseq, hiseq2500, hiseqX) | |
--sequencing-center <SEQUENCING_CENTER> | The sequencing center from which the data originated | |
--predicted-insert-size <PREDICTED_INSERT_SIZE> | Predicted median insert size, to insert into the read group header | |
--description <DESCRIPTION> | Description of the read group | |
--comment <COMMENT> | Comment(s) to include in the output file’s header | |
--run-date <RUN_DATE> | Date the run was produced, to insert into the read group header | |
--threads <THREADS> | Number of threads for the multi-threaded pipeline | |
--compression-level <COMPRESSION_LEVEL> | Compression level for output BAM (0-12) | 1 |
--scheduler <SCHEDULER> | Scheduler strategy for thread work assignment | balanced-chase-drain |
--pipeline-stats <PIPELINE_STATS> | Print detailed pipeline statistics at completion | false |
--deadlock-timeout <DEADLOCK_TIMEOUT> | Timeout in seconds for deadlock detection (default: 10, 0 = disabled) | 10 |
--deadlock-recover <DEADLOCK_RECOVER> | Enable automatic deadlock recovery (default: false, detection only) | false |
--queue-memory <QUEUE_MEMORY> | Pipeline queue memory limit per thread (default) or total | 768 |
--queue-memory-per-thread <QUEUE_MEMORY_PER_THREAD> | Interpret –queue-memory as per-thread (true, default) or total (false) | true |
--queue-memory-limit-mb <QUEUE_MEMORY_LIMIT_MB> | DEPRECATED: Use –queue-memory instead. Memory limit for pipeline queues in megabytes | |
--async-reader <ASYNC_READER> | Wrap FASTQ inputs in a userspace async prefetch reader. Dedicates one OS thread per input stream to issue reads ahead of decompression/parsing. Hidden experimental flag | false |
correct
Category: UMI EXTRACTION
Correct UMIs in a BAM file to a fixed set of UMIs
Description
Corrects UMIs stored in BAM files when a set of fixed UMIs is in use.
If the set of UMIs used in an experiment is known and is a subset of the possible randomers
of the same length, it is possible to error-correct UMIs prior to grouping reads by UMI. This
tool takes an input BAM with UMIs in the RX tag and set of known UMIs (either on
the command line or in a file) and produces:
- A new BAM with corrected UMIs written to the
RXtag - Optionally a set of metrics about the representation of each UMI in the set
- Optionally a second BAM file of reads whose UMIs could not be corrected within the specific parameters
All of the fixed UMIs must be of the same length, and all UMIs in the BAM file must also have
the same length. Multiple UMIs that are concatenated with hyphens (e.g. AACCAGT-AGGTAGA) are
split apart, corrected individually and then re-assembled. A read is accepted only if all the
UMIs can be corrected.
Correction Parameters
Correction is controlled by two parameters that are applied per-UMI:
- –max-mismatches controls how many mismatches (no-calls are counted as mismatches) are tolerated between a UMI as read and a fixed UMI
- –min-distance controls how many more mismatches the next best hit must have
For example, with two fixed UMIs AAAAA and CCCCC and --max-mismatches=3 and --min-distance=2:
- AAAAA would match to AAAAA
- AAGTG would match to AAAAA with three mismatches because CCCCC has six mismatches and 6 >= 3 + 2
- AACCA would be rejected because it is 2 mismatches to AAAAA and 3 to CCCCC and 3 <= 2 + 2
Specifying UMIs
The set of fixed UMIs may be specified on the command line using --umis umi1 umi2 ... or via
one or more files of UMIs with a single sequence per line using --umi-files umis.txt more_umis.txt.
If there are multiple UMIs per template, leading to hyphenated UMI tags, the values for the fixed
UMIs should be single, non-hyphenated UMIs (e.g. if a record has RX:Z:ACGT-GGCA, you would use
--umis ACGT GGCA).
Original UMI Storage
Records which have their UMIs corrected (i.e. the UMI is not identical to one of the expected
UMIs but is close enough to be corrected) will by default have their original UMI stored in the
OX tag. This can be disabled with the --dont-store-original-umis option.
Arguments
| Flag | Description | Default |
|---|---|---|
-i, --input <INPUT> | Input BAM file | required |
-o, --output <OUTPUT> | Output BAM file | required |
--async-reader <ASYNC_READER> | Enable async userspace prefetch on the input BAM | false |
-r, --rejects <REJECTS> | Optional output BAM file for rejected reads | |
-M, --metrics <METRICS> | Optional output path for metrics TSV file | |
--max-mismatches <MAX_MISMATCHES> | Maximum number of mismatches allowed | 2 |
-d, --min-distance <MIN_DISTANCE_DIFF> | Minimum difference between best and second-best match | required |
-u, --umis <UMIS> | Fixed UMI sequences (can be specified multiple times) | |
-U, --umi-files <UMI_FILES> | Files containing UMI sequences, one per line | |
--dont-store-original-umis <DONT_STORE_ORIGINAL_UMIS> | Don’t store original UMIs in a separate tag | false |
--cache-size <CACHE_SIZE> | Size of the LRU cache for UMI matching | 100000 |
--min-corrected <MIN_CORRECTED> | Minimum fraction of reads that must pass correction | |
--revcomp <REVCOMP> | Reverse complement UMIs before matching | false |
--threads <THREADS> | Number of threads for the multi-threaded pipeline | |
--compression-level <COMPRESSION_LEVEL> | Compression level for output BAM (0-12) | 1 |
--scheduler <SCHEDULER> | Scheduler strategy for thread work assignment | balanced-chase-drain |
--pipeline-stats <PIPELINE_STATS> | Print detailed pipeline statistics at completion | false |
--deadlock-timeout <DEADLOCK_TIMEOUT> | Timeout in seconds for deadlock detection (default: 10, 0 = disabled) | 10 |
--deadlock-recover <DEADLOCK_RECOVER> | Enable automatic deadlock recovery (default: false, detection only) | false |
--queue-memory <QUEUE_MEMORY> | Pipeline queue memory limit per thread (default) or total | 768 |
--queue-memory-per-thread <QUEUE_MEMORY_PER_THREAD> | Interpret –queue-memory as per-thread (true, default) or total (false) | true |
--queue-memory-limit-mb <QUEUE_MEMORY_LIMIT_MB> | DEPRECATED: Use –queue-memory instead. Memory limit for pipeline queues in megabytes |
fastq
Category: ALIGNMENT
Convert BAM to FASTQ format
Description
Convert a BAM file to interleaved FASTQ format.
Reads BAM records and outputs FASTQ to stdout for piping to aligners. Input should be queryname-sorted or template-coordinate sorted.
EXAMPLES:
Pipe to bwa mem for alignment
fgumi fastq -i unmapped.bam | bwa mem -t 16 -p -K 150000000 -Y ref.fa -
With multi-threaded BAM decompression
fgumi fastq -i unmapped.bam -@ 4 | bwa mem -t 16 -p ref.fa -
Exclude secondary and supplementary alignments (default)
fgumi fastq -i aligned.bam -F 0x900 | bwa mem …
Arguments
| Flag | Description | Default |
|---|---|---|
-i, --input <INPUT> | Input BAM file | required |
-o, --output <OUTPUT> | Output FASTQ file. If omitted, the FASTQ stream is written to stdout (the default, intended for piping straight to an aligner) | |
-n, --no-read-suffix <NO_SUFFIX> | Don’t append /1 and /2 to read names | false |
-F, --exclude-flags <EXCLUDE_FLAGS> | Exclude reads with any of these flags present [0x900 = secondary|supplementary] | 2304 |
-f, --require-flags <REQUIRE_FLAGS> | Only include reads with all of these flags present | 0 |
-@, --threads <THREADS> | Number of threads for BAM decompression | 1 |
-K, --bwa-chunk-size <BWA_CHUNK_SIZE> | BWA -K parameter value (bases per batch). Sizes output buffer to match bwa’s batch size for optimal pipe throughput. Default matches common bwa mem usage | 150000000 |
zipper
Category: ALIGNMENT
Zip unmapped BAM with aligned BAM
Description
Merges unmapped and mapped BAM files, transferring tags and metadata.
Takes an unmapped BAM (typically from FASTQ) and a mapped BAM (after alignment) and merges them, copying tags from the unmapped to mapped reads. Both BAMs must be queryname sorted or grouped, and have the same read name ordering.
The tool transfers tags from the unmapped reads to their corresponding mapped reads. For reads mapped to the negative strand, tags can be optionally reversed or reverse-complemented. All QC pass/fail flags are also transferred from the unmapped to mapped reads.
Tag Manipulation
You can specify which tags to manipulate for reads mapped to the negative strand:
- –tags-to-reverse: Reverses array and string tags (e.g., [1,2,3] becomes [3,2,1])
- –tags-to-revcomp: Reverse complements sequence tags (e.g., AGAGG becomes CCTCT)
Named tag sets like “Consensus” are automatically expanded to their constituent tags:
- Consensus: aD bD cD aM bM cM aE bE cE ad bd cd ae be ce ac bc
Default Behavior
By default, input is read from stdin and output is written to stdout, allowing for streaming workflows like:
Recommended when the aligner can emit uncompressed BAM:
bwa-mem3 mem –bam=0 -t 16 -p -K 150000000 -Y ref.fa reads.fq | fgumi zipper -u unmapped.bam -r ref.fa | fgumi sort -i /dev/stdin -o output.bam –order template-coordinate
SAM-only aligners (e.g. classic bwa mem, bwa-mem2):
bwa mem -t 16 -p -K 150000000 -Y ref.fa reads.fq | fgumi zipper -u unmapped.bam -r ref.fa | fgumi sort -i /dev/stdin -o output.bam –order template-coordinate
Uncompressed BAM avoids the SAM text formatting/parsing round-trip in both processes and adds only ~26 bytes of BGZF framing per ~64 KiB block. Compressed BAM on a pipe is not recommended — it burns CPU on the writer and reader for data the sort step will re-compress anyway.
Arguments
| Flag | Description | Default |
|---|---|---|
-i, --input <INPUT> | Input mapped SAM or BAM file (or - for stdin; SAM or BAM is auto-detected). For streaming pipelines, uncompressed BAM (e.g. bwa-mem3 mem --bam=0) is the fastest option — it skips both SAM text formatting on the aligner side and SAM parsing on this side. SAM is fine if your aligner can’t emit BAM. Compressed BAM on a pipe wastes CPU on both ends | - |
-u, --unmapped <UNMAPPED> | Input unmapped BAM file containing original tags | required |
-r, --reference <REFERENCE> | Reference FASTA file (must have accompanying .dict file) | required |
-o, --output <OUTPUT> | Output BAM file (or - for stdout) | - |
--tags-to-remove <TAGS_TO_REMOVE> | Tags to remove from mapped reads before copying unmapped tags | |
--tags-to-reverse <TAGS_TO_REVERSE> | Tags to reverse for reads mapped to negative strand | |
--tags-to-revcomp <TAGS_TO_REVCOMP> | Tags to reverse complement for reads mapped to negative strand | |
-b, --buffer <BUFFER> | Buffer size for template channel (default: 50000) | 50000 |
-t, --threads <THREADS> | Number of threads to use for processing (default: 1, single-threaded) | 1 |
--compression-level <COMPRESSION_LEVEL> | Compression level for output BAM (0-12) | 1 |
-K, --bwa-chunk-size <BWA_CHUNK_SIZE> | BWA -K parameter value (bases per batch). Used to optimize buffer sizing for stdin input. The buffer grows adaptively based on observed bytes per batch. Default matches common bwa mem usage | 150000000 |
--exclude-missing-reads <EXCLUDE_MISSING_READS> | Exclude reads from the unmapped BAM that are not present in the aligned BAM. Useful when reads were intentionally removed (e.g., by adapter trimming) prior to alignment | false |
--skip-pa-tags <SKIP_TC_TAGS> | Skip adding pa (primary alignment) tags to secondary/supplementary reads. By default, zipper adds a pa tag containing the primary alignment’s template sort key coordinates, which enables correct template-coordinate sorting and deduplication of these reads. Use this flag if you don’t need this functionality | false |
--restore-unconverted-bases <RESTORE_UNCONVERTED_BASES> | Restore unconverted bases in EM-seq consensus reads after bwameth re-alignment | false |
sort
Category: ALIGNMENT
Sort BAM file by coordinate, queryname, or template-coordinate
Description
Sort a BAM file using high-performance external merge-sort.
This tool provides efficient BAM sorting with support for multiple sort orders:
SORT ORDERS:
coordinate Standard genomic coordinate sort (tid → pos → strand).
Use for IGV visualization, variant calling, fgumi review.
queryname Lexicographic read name sort (fast, default sub-sort).
queryname::lex Short alias for lexicographic ordering (same as above).
queryname::lexicographic Explicit lexicographic ordering (same as above).
queryname::natural Natural numeric ordering (samtools-compatible).
Use for fgumi zipper, template-level operations.
template-coordinate Template-level position sort for UMI grouping.
Use for fgumi group, fgumi dedup, and fgumi downsample input.
PERFORMANCE:
- 1.9x faster than samtools on template-coordinate sort
- Handles BAM files larger than available RAM via spill-to-disk
- Uses parallel sorting (–threads) for in-memory chunks
- Configurable temp file compression (–temp-compression)
- Default 768M per-thread memory limit (samtools-compatible); pass
--max-memory autoto detect system memory (opt-in)
EXAMPLES:
Sort for fgumi group input
fgumi sort -i aligned.bam -o sorted.bam –order template-coordinate
Sort by coordinate for IGV
fgumi sort -i input.bam -o sorted.bam –order coordinate
Sort by queryname for zipper
fgumi sort -i input.bam -o sorted.bam –order queryname
Multi-threaded sort (default 768M per thread)
fgumi sort -i input.bam -o sorted.bam –order template-coordinate –threads 8
Override the per-thread memory limit
fgumi sort -i input.bam -o sorted.bam -m 2GiB –threads 8
Opt in to auto-detected system memory (subtracts –memory-reserve)
fgumi sort -i input.bam -o sorted.bam -m auto –threads 8
Reserve extra memory for bwa mem running in a pipeline
fgumi sort -i input.bam -o sorted.bam –memory-reserve 12GiB –threads 4
Verify a BAM file is correctly sorted
fgumi sort -i sorted.bam –verify –order template-coordinate
Spread spill chunks across multiple temp dirs (round-robin, free-space aware)
fgumi sort -i in.bam -o out.bam -T /mnt/ssd1 -T /mnt/ssd2
Same via FGUMI_TMP_DIRS env var (PATH-style list)
FGUMI_TMP_DIRS=/mnt/ssd1:/mnt/ssd2 fgumi sort -i in.bam -o out.bam
Arguments
| Flag | Description | Default |
|---|---|---|
-i, --input <INPUT> | Input BAM file | required |
-o, --output <OUTPUT> | Output BAM file (required unless –verify is used) | |
--verify <VERIFY> | Verify the input file is correctly sorted (no output written) | false |
--order <ORDER> | Sort order | template-coordinate |
--key-types <KEY_TYPES> | Which optional lanes to keep in the template-coordinate sort key | |
-m, --max-memory <MAX_MEMORY> | Maximum memory for in-memory sorting | 768M |
--memory-reserve <MEMORY_RESERVE> | Memory to reserve for other processes when –max-memory=auto | auto |
--memory-per-thread <MEMORY_PER_THREAD> | Scale memory limit by thread count (samtools behavior) | true |
-T, --tmp-dir <TMP_DIRS> | Temporary directory for intermediate files. Repeatable | |
-@, --threads <THREADS> | Number of threads for parallel operations | 1 |
--compression-level <COMPRESSION_LEVEL> | Compression level for output BAM (0-12) | 1 |
--temp-compression <TEMP_COMPRESSION> | Compression level for temporary chunk files (0-9) | 1 |
--temp-codec <TEMP_CODEC> | Codec used for temporary spill chunks: zstd (default) or bgzf | zstd |
--write-index <WRITE_INDEX> | Write BAM index (.bai) alongside output | false |
--async-reader <ASYNC_READER> | Enable async userspace prefetch on the input BAM | false |
merge
Category: ALIGNMENT
Merge pre-sorted BAM files into a single sorted BAM
Description
Merge pre-sorted BAM files into a single sorted BAM.
Performs a k-way merge of multiple BAM files that are already sorted in the
same order, producing a single merged output that preserves the sort order.
Similar to samtools merge, but supports template-coordinate order.
Input files must all be sorted in the specified sort order.
EXAMPLES:
Merge coordinate-sorted BAMs
fgumi merge -o merged.bam sorted1.bam sorted2.bam sorted3.bam
Merge template-coordinate sorted BAMs
fgumi merge -o merged.bam –order template-coordinate tc1.bam tc2.bam
Merge from a file listing input BAMs (one per line)
fgumi merge -o merged.bam -b input_list.txt –order queryname
Merge with multiple threads
fgumi merge -o merged.bam -@ 4 sorted1.bam sorted2.bam
Arguments
| Flag | Description | Default |
|---|---|---|
-o, --output <OUTPUT> | Output BAM file | required |
inputs <INPUTS> | Input BAM files to merge (positional) | |
-b, --input-list <INPUT_LIST> | File containing a list of input BAM paths, one per line | |
--order <ORDER> | Sort order of the input files | template-coordinate |
-@, --threads <THREADS> | Number of threads for parallel operations | 1 |
--compression-level <COMPRESSION_LEVEL> | Compression level for output BAM (1-12) | 6 |
group
Category: GROUP
Group reads by UMI to identify reads from the same original molecule
Description
Groups reads together that appear to have come from the same original molecule. Reads are grouped by template, and then templates are sorted by the 5’ mapping positions of the reads from the template, used from earliest mapping position to latest. Reads that have the same end positions are then sub-grouped by UMI sequence.
Requires input to be template-coordinate sorted (header must advertise
SO:unsorted, GO:query, and SS:template-coordinate). Sort upstream sources
(fgumi extract, samtools sort -n, fgumi merge --order queryname, etc.)
with fgumi sort -i input.bam -o sorted.bam --order template-coordinate
before piping into this tool. Output is always written in template-coordinate
order, sorted by:
- The lower genome coordinate of the two outer ends of the templates (strand-aware)
- The sequencing library
- The cell barcode (CB tag, if present)
- The assigned UMI tag
- Read Name
During grouping, reads and templates are filtered out as follows:
- Templates are filtered if all reads for the template are unmapped
- Templates are filtered if any non-secondary, non-supplementary read has mapping quality < min-map-q
- Templates are filtered if any UMI sequence contains one or more N bases
- Templates are filtered if –min-umi-length is specified and the UMI does not meet the length requirement
- Records are filtered out if flagged as either secondary or supplementary
Grouping of UMIs is performed by one of four strategies:
- identity: only reads with identical UMI sequences are grouped together. This strategy may be useful for evaluating data, but should generally be avoided as it will generate multiple UMI groups per original molecule in the presence of errors.
- edit: reads are clustered into groups such that each read within a group has at least one other read in the group with <= edits differences and there are inter-group pairings with <= edits differences. Effective when there are small numbers of reads per UMI, but breaks down at very high coverage of UMIs.
- adjacency: a version of the directed adjacency method described in umi_tools (http://dx.doi.org/10.1101/051755) that allows for errors between UMIs but only when there is a count gradient.
- paired: similar to adjacency but for methods that produce templates such that a read with A-B is related to but not identical to a read with B-A. Expects the UMI sequences to be stored in a single SAM tag separated by a hyphen (e.g. ACGT-CCGG) and allows for one of the two UMIs to be absent (e.g. ACGT- or -ACGT). The molecular IDs produced have more structure than for single UMI strategies and are of the form {base}/{A|B}. E.g. two UMI pairs would be mapped as follows: AAAA-GGGG -> 1/A, GGGG-AAAA -> 1/B.
Strategies edit, adjacency, and paired make use of the –edits parameter to control the matching of non-identical UMIs.
By default, all UMIs must be the same length. If –min-umi-length=len is specified then reads that have a UMI shorter than len will be discarded, and when comparing UMIs of different lengths, the first len bases will be compared, where len is the length of the shortest UMI. The UMI length is the number of [ACGT] bases in the UMI (i.e. does not count dashes and other non-ACGT characters). This option is not implemented for reads with UMI pairs (i.e. using the paired assigner).
Note: the –min-map-q parameter defaults to 0 in duplicate marking mode and 1 otherwise, and is directly settable on the command line.
Cell Barcodes
If the input data contains cell barcodes (e.g. from single-cell sequencing), reads at the same
genomic position are partitioned by cell barcode before UMI grouping. This ensures that reads from
different cells are never grouped together, even if they share a UMI sequence and mapping position.
The cell barcode is read from the standard CB tag. No correction or
error-handling is performed on cell barcodes; they must be corrected upstream.
Multi-threaded operation is supported via –threads N, which spawns N pipeline threads allocated based on the command’s workload profile to optimize performance.
Example: –threads 8 spawns 8 pipeline threads (2 reader, 4 workers, 2 writer)
Note: when –parallel-group-min-templates (or –allow-unmapped) engages the parallel UMI assigner, each parallel assigner constructs its own rayon thread pool of size –threads, independent of the pipeline threads above. As an example, one pipeline worker overlapping a single parallel assigner briefly runs ~2 * –threads OS threads; this is not an upper bound, because multiple pipeline workers can each spawn a –threads-sized pool concurrently and push the live thread count higher still. See –parallel-group-min-templates for details.
Arguments
| Flag | Description | Default |
|---|---|---|
-i, --input <INPUT> | Input BAM file | required |
-o, --output <OUTPUT> | Output BAM file | required |
--async-reader <ASYNC_READER> | Enable async userspace prefetch on the input BAM | false |
-f, --family-size-histogram <FAMILY_SIZE_HISTOGRAM> | Optional output of tag family size counts | |
-g, --grouping-metrics <GROUPING_METRICS> | Optional output of UMI grouping metrics | |
-M, --metrics <METRICS> | Output prefix for all group metrics files | |
-m, --min-map-q <MIN_MAP_Q> | Minimum mapping quality for mapped reads | |
-n, --include-non-pf-reads <INCLUDE_NON_PF_READS> | Include non-PF reads | false |
--allow-unmapped <ALLOW_UNMAPPED> | Allow fully unmapped templates (both reads unmapped). Input must be template-coordinate sorted (fgumi sort --order template-coordinate) | false |
| `–parallel-group-min-templates <N | auto>` | Enable the parallel UMI assigner for position groups with at least this many templates. Useful for amplicon and other workflows where individual mapped position groups are very large; the default for normal whole-genome data is to stay sequential. Has an effect only when --threads is greater than 1: with --threads 1 the assigner always falls back to the sequential implementation |
-s, --strategy <STRATEGY> | The UMI assignment strategy | required |
-e, --edits <EDITS> | The allowable number of edits between UMIs | 1 |
-l, --min-umi-length <MIN_UMI_LENGTH> | The minimum UMI length | |
--threads <THREADS> | Number of threads for the multi-threaded pipeline | |
--compression-level <COMPRESSION_LEVEL> | Compression level for output BAM (0-12) | 1 |
--index-threshold <INDEX_THRESHOLD> | Minimum UMIs per position to use N-gram/BK-tree index for faster grouping. Set to 0 to always use linear scan. Only affects Adjacency/Paired strategies | 100 |
--no-umi <NO_UMI> | Skip UMI-based grouping; group by position only. Forces identity strategy and ignores any existing UMI tags | false |
--scheduler <SCHEDULER> | Scheduler strategy for thread work assignment | balanced-chase-drain |
--pipeline-stats <PIPELINE_STATS> | Print detailed pipeline statistics at completion | false |
--deadlock-timeout <DEADLOCK_TIMEOUT> | Timeout in seconds for deadlock detection (default: 10, 0 = disabled) | 10 |
--deadlock-recover <DEADLOCK_RECOVER> | Enable automatic deadlock recovery (default: false, detection only) | false |
--queue-memory <QUEUE_MEMORY> | Pipeline queue memory limit per thread (default) or total | 768 |
--queue-memory-per-thread <QUEUE_MEMORY_PER_THREAD> | Interpret –queue-memory as per-thread (true, default) or total (false) | true |
--queue-memory-limit-mb <QUEUE_MEMORY_LIMIT_MB> | DEPRECATED: Use –queue-memory instead. Memory limit for pipeline queues in megabytes |
dedup
Category: DEDUP
Mark or remove PCR duplicates using UMI information
Description
Marks or removes PCR duplicates from a BAM file using UMI information.
Requires template-coordinate sorted input with tc tags on secondary/supplementary
reads (added by fgumi zipper).
Within each UMI family, the template with the highest sum of base qualities is selected as the representative; all others are marked as duplicates.
Input Requirements
- Must be processed with
fgumi zipper(addstctag for secondary/supplementary reads) - Must be sorted with
fgumi sort --order template-coordinate - UMI tags on reads (RX tag), unless
--no-umiis specified
Note: Using samtools sort will NOT work correctly because it doesn’t use the
tc tag for template-coordinate ordering of secondary/supplementary reads.
Output Modes
- Mark only (default): Set duplicate flag (0x400) on non-representative reads
- Remove (–remove-duplicates): Exclude duplicate reads from output entirely
Cell Barcodes
If the input data contains cell barcodes (e.g. from single-cell sequencing), reads at the same
genomic position are partitioned by cell barcode before deduplication. This ensures that reads from
different cells are never marked as duplicates of each other, even if they share a UMI sequence and
mapping position. The cell barcode is read from the standard CB tag. No
correction or error-handling is performed on cell barcodes; they must be corrected upstream.
Arguments
| Flag | Description | Default |
|---|---|---|
-i, --input <INPUT> | Input BAM file | required |
-o, --output <OUTPUT> | Output BAM file | required |
--async-reader <ASYNC_READER> | Enable async userspace prefetch on the input BAM | false |
-m, --metrics <METRICS> | Path to write deduplication metrics | |
-H, --family-size-histogram <FAMILY_SIZE_HISTOGRAM> | Path to write family size histogram | |
-r, --remove-duplicates <REMOVE_DUPLICATES> | Remove duplicates instead of just marking them | false |
-q, --min-map-q <MIN_MAP_Q> | Minimum mapping quality for a read to be included | |
-n, --include-non-pf-reads <INCLUDE_NON_PF_READS> | Include reads flagged as not passing QC | false |
-s, --strategy <STRATEGY> | UMI grouping strategy | adjacency |
-e, --edits <EDITS> | Maximum edit distance for UMI grouping | 1 |
-l, --min-umi-length <MIN_UMI_LENGTH> | Minimum UMI length (UMIs shorter than this are discarded) | |
--threads <THREADS> | Number of threads for the multi-threaded pipeline | |
--compression-level <COMPRESSION_LEVEL> | Compression level for output BAM (0-12) | 1 |
--index-threshold <INDEX_THRESHOLD> | Minimum UMIs per position to use index for faster grouping | 100 |
--no-umi <NO_UMI> | Skip UMI-based grouping; group by position only. Forces identity strategy and ignores any existing UMI tags | false |
--scheduler <SCHEDULER> | Scheduler strategy for thread work assignment | balanced-chase-drain |
--pipeline-stats <PIPELINE_STATS> | Print detailed pipeline statistics at completion | false |
--deadlock-timeout <DEADLOCK_TIMEOUT> | Timeout in seconds for deadlock detection (default: 10, 0 = disabled) | 10 |
--deadlock-recover <DEADLOCK_RECOVER> | Enable automatic deadlock recovery (default: false, detection only) | false |
--queue-memory <QUEUE_MEMORY> | Pipeline queue memory limit per thread (default) or total | 768 |
--queue-memory-per-thread <QUEUE_MEMORY_PER_THREAD> | Interpret –queue-memory as per-thread (true, default) or total (false) | true |
--queue-memory-limit-mb <QUEUE_MEMORY_LIMIT_MB> | DEPRECATED: Use –queue-memory instead. Memory limit for pipeline queues in megabytes |
simplex
Category: CONSENSUS
Call simplex consensus sequences from UMI-grouped reads
Description
Calls consensus sequences from reads with the same unique molecular tag.
Reads with the same unique molecular tag are examined base-by-base to assess the likelihood of each base in the source molecule. The likelihood model is as follows:
- First, the base qualities are adjusted. The base qualities are assumed to represent the probability of a sequencing error (i.e. the sequencer observed the wrong base present on the cluster/flowcell/well). The base quality scores are converted to probabilities incorporating a probability representing the chance of an error from the time the unique molecular tags were integrated to just prior to sequencing. The resulting probability is the error rate of all processes from right after integrating the molecular tag through to the end of sequencing.
- Next, a consensus sequence is called for all reads with the same unique molecular tag base-by-base. For a given base position in the reads, the likelihoods that an A, C, G, or T is the base for the underlying source molecule respectively are computed by multiplying the likelihood of each read observing the base position being considered. The probability of error (from 1.) is used when the observed base does not match the hypothesized base for the underlying source molecule, while one minus that probability is used otherwise. The computed likelihoods are normalized by dividing them by the sum of all four likelihoods to produce a posterior probability, namely the probability that the source molecule was an A, C, G, or T from just after integrating molecular tag through to sequencing, given the observations. The base with the maximum posterior probability as the consensus call, and the posterior probability is used as its raw base quality.
- Finally, the consensus raw base quality is modified by incorporating the probability of an error prior to integrating the unique molecular tags. Therefore, the probability used for the final consensus base quality is the posterior probability of the source molecule having the consensus base given the observed reads with the same molecular tag, all the way from sample extraction and through sample and library preparation, through preparing the library for sequencing (e.g. amplification, target selection), and finally, through sequencing.
This tool assumes that reads with the same tag are grouped together (consecutive in the file). Also, this tool calls each end of a pair independently, and does not jointly call bases that overlap within a pair. Insertion or deletion errors in the reads are not considered in the consensus model.
The consensus reads produced are unaligned, due to the difficulty and error-prone nature of inferring the consensus alignment. Consensus reads should therefore be aligned after, which should not be too expensive as likely there are far fewer consensus reads than input raw reads.
Particular attention should be paid to setting the –min-reads parameter as this can have a dramatic effect on both results and runtime. For libraries with low duplication rates (e.g. 100-300X exomes libraries) in which it is desirable to retain singleton reads while making consensus reads from sets of duplicates, –min-reads=1 is appropriate. For libraries with high duplication rates where it is desirable to only produce consensus reads supported by 2+ reads to allow error correction, –min-reads=2 or higher is appropriate. After generation, consensus reads can be further filtered using the filter tool. As such it is always safe to run with –min-reads=1 and filter later, but filtering at this step can improve performance significantly.
Consensus reads have a number of additional optional tags set in the resulting BAM file. The tags break down into those that are single-valued per read:
consensus depth [cD] (int) : the maximum depth of raw reads at any point in the consensus read consensus min depth [cM] (int) : the minimum depth of raw reads at any point in the consensus read consensus error rate [cE] (float): the fraction of bases in raw reads disagreeing with the final consensus calls
And those that have a value per base:
consensus depth [cd] (short[]): the count of bases contributing to the consensus read at each position consensus errors [ce] (short[]): the number of bases from raw reads disagreeing with the final consensus base
The per base depths and errors are both capped at 32,767. In all cases no-calls (Ns) and bases below the –min-input-base-quality are not counted in tag value calculations.
Arguments
| Flag | Description | Default |
|---|---|---|
-i, --input <INPUT> | Input BAM file | required |
-o, --output <OUTPUT> | Output BAM file | required |
--async-reader <ASYNC_READER> | Enable async userspace prefetch on the input BAM | false |
-r, --rejects <REJECTS> | Optional output BAM file for rejected reads | |
-s, --stats <STATS> | Optional output file for statistics | |
-p, --read-name-prefix <READ_NAME_PREFIX> | Prefix for consensus read names | |
-R, --read-group-id <READ_GROUP_ID> | Read group ID for consensus reads | A |
-1, --error-rate-pre-umi <ERROR_RATE_PRE_UMI> | Phred-scaled error rate prior to UMI integration | 45 |
-2, --error-rate-post-umi <ERROR_RATE_POST_UMI> | Phred-scaled error rate post UMI integration | 40 |
-m, --min-input-base-quality <MIN_INPUT_BASE_QUALITY> | Minimum base quality in raw reads to use for consensus | 10 |
-B, --output-per-base-tags <OUTPUT_PER_BASE_TAGS> | Produce per-base tags (cd, ce) in addition to per-read tags | true |
--trim <TRIM> | Quality-trim reads before consensus calling (removes low-quality bases from ends) | false |
--min-consensus-base-quality <MIN_CONSENSUS_BASE_QUALITY> | Minimum consensus base quality (output consensus bases below this are masked to N) | 2 |
--consensus-call-overlapping-bases <CONSENSUS_CALL_OVERLAPPING_BASES> | Consensus call overlapping bases in read pairs before UMI consensus calling | true |
--threads <THREADS> | Number of threads for the multi-threaded pipeline | |
--compression-level <COMPRESSION_LEVEL> | Compression level for output BAM (0-12) | 1 |
-M, --min-reads <MIN_READS> | Minimum number of reads to produce a consensus (required, no default) Matches fgbio’s CallMolecularConsensusReads which requires this argument | required |
--max-reads <MAX_READS> | Maximum reads to use per tag family (downsample if exceeded) | |
--scheduler <SCHEDULER> | Scheduler strategy for thread work assignment | balanced-chase-drain |
--pipeline-stats <PIPELINE_STATS> | Print detailed pipeline statistics at completion | false |
--deadlock-timeout <DEADLOCK_TIMEOUT> | Timeout in seconds for deadlock detection (default: 10, 0 = disabled) | 10 |
--deadlock-recover <DEADLOCK_RECOVER> | Enable automatic deadlock recovery (default: false, detection only) | false |
--queue-memory <QUEUE_MEMORY> | Pipeline queue memory limit per thread (default) or total | 768 |
--queue-memory-per-thread <QUEUE_MEMORY_PER_THREAD> | Interpret –queue-memory as per-thread (true, default) or total (false) | true |
--queue-memory-limit-mb <QUEUE_MEMORY_LIMIT_MB> | DEPRECATED: Use –queue-memory instead. Memory limit for pipeline queues in megabytes | |
--methylation-mode <METHYLATION_MODE> | Methylation-aware consensus calling mode. EM-Seq: C→T at ref-C = unmethylated (enzymatic conversion); TAPs: C→T at ref-C = methylated. Emits MM/ML methylation tags and cu/ct per-base count tags on consensus reads. Requires –ref | |
--ref <REFERENCE> | Path to the reference FASTA file (required when –methylation-mode is set) |
duplex
Category: CONSENSUS
Call duplex consensus sequences from UMI-grouped reads
Description
Calls duplex consensus sequences from reads generated from the same double-stranded source molecule. Prior
to running this tool, reads must have been grouped with group using the paired strategy. Doing
so will apply (by default) MI tags to all reads of the form */A and */B where the /A and /B suffixes
with the same identifier denote reads that are derived from opposite strands of the same source duplex molecule.
Reads from the same unique molecule are first partitioned by source strand and assembled into single strand consensus molecules as described by the simplex command. Subsequently, for molecules that have at least one observation of each strand, duplex consensus reads are assembled by combining the evidence from the two single strand consensus reads.
Because of the nature of duplex sequencing, this tool does not support fragment reads - if found in the input they are ignored. Similarly, read pairs for which consensus reads cannot be generated for one or other read (R1 or R2) are omitted from the output.
The consensus reads produced are unaligned, due to the difficulty and error-prone nature of inferring the consensus alignment. Consensus reads should therefore be aligned after, which should not be too expensive as likely there are far fewer consensus reads than input raw reads.
Consensus reads have a number of additional optional tags set in the resulting BAM file. The tag names follow a pattern where the first letter (a, b or c) denotes that the tag applies to the first single strand consensus (a), second single-strand consensus (b) or the final duplex consensus (c). The second letter is intended to capture the meaning of the tag (e.g. d=depth, m=min depth, e=errors/error-rate) and is upper case for values that are one per read and lower case for values that are one per base.
The tags break down into those that are single-valued per read:
consensus depth [aD,bD,cD] (int) : the maximum depth of raw reads at any point in the consensus reads consensus min depth [aM,bM,cM] (int) : the minimum depth of raw reads at any point in the consensus reads consensus error rate [aE,bE,cE] (float): the fraction of bases in raw reads disagreeing with the final consensus calls
And those that have a value per base (duplex values are not generated, but can be generated by summing):
consensus depth [ad,bd] (short[]): the count of bases contributing to each single-strand consensus read at each position consensus errors [ae,be] (short[]): the count of bases from raw reads disagreeing with the final single-strand consensus base consensus bases [ac,bc] (string) : the single-strand consensus bases consensus quals [aq,bq] (string) : the single-strand consensus qualities
The per base depths and errors are both capped at 32,767. In all cases no-calls (Ns) and bases below the min-input-base-quality are not counted in tag value calculations.
The –min-reads option can take 1-3 values similar to filter. For example:
fgumi duplex … –min-reads 10,5,3
If fewer than three values are supplied, the last value is repeated (i.e. 5,4 -> 5 4 4 and 1 -> 1 1 1). The
first value applies to the final consensus read, the second value to one single-strand consensus, and the last
value to the other single-strand consensus. It is required that if values two and three differ,
the more stringent value comes earlier.
Arguments
| Flag | Description | Default |
|---|---|---|
-i, --input <INPUT> | Input BAM file | required |
-o, --output <OUTPUT> | Output BAM file | required |
--async-reader <ASYNC_READER> | Enable async userspace prefetch on the input BAM | false |
-r, --rejects <REJECTS> | Optional output BAM file for rejected reads | |
-s, --stats <STATS> | Optional output file for statistics | |
-p, --read-name-prefix <READ_NAME_PREFIX> | Prefix for consensus read names | |
-R, --read-group-id <READ_GROUP_ID> | Read group ID for consensus reads | A |
-1, --error-rate-pre-umi <ERROR_RATE_PRE_UMI> | Phred-scaled error rate prior to UMI integration | 45 |
-2, --error-rate-post-umi <ERROR_RATE_POST_UMI> | Phred-scaled error rate post UMI integration | 40 |
-m, --min-input-base-quality <MIN_INPUT_BASE_QUALITY> | Minimum base quality in raw reads to use for consensus | 10 |
-B, --output-per-base-tags <OUTPUT_PER_BASE_TAGS> | Produce per-base tags (cd, ce) in addition to per-read tags | true |
--trim <TRIM> | Quality-trim reads before consensus calling (removes low-quality bases from ends) | false |
--min-consensus-base-quality <MIN_CONSENSUS_BASE_QUALITY> | Minimum consensus base quality (output consensus bases below this are masked to N) | 2 |
--consensus-call-overlapping-bases <CONSENSUS_CALL_OVERLAPPING_BASES> | Consensus call overlapping bases in read pairs before UMI consensus calling | true |
--threads <THREADS> | Number of threads for the multi-threaded pipeline | |
--compression-level <COMPRESSION_LEVEL> | Compression level for output BAM (0-12) | 1 |
-M, --min-reads <MIN_READS> | Minimum reads for consensus calling. Can specify 1-3 values: [duplex] or [duplex, AB/BA] or [duplex, AB, BA] | 1 |
--max-reads-per-strand <MAX_READS_PER_STRAND> | Maximum reads per strand (downsample if exceeded) | |
--scheduler <SCHEDULER> | Scheduler strategy for thread work assignment | balanced-chase-drain |
--pipeline-stats <PIPELINE_STATS> | Print detailed pipeline statistics at completion | false |
--deadlock-timeout <DEADLOCK_TIMEOUT> | Timeout in seconds for deadlock detection (default: 10, 0 = disabled) | 10 |
--deadlock-recover <DEADLOCK_RECOVER> | Enable automatic deadlock recovery (default: false, detection only) | false |
--queue-memory <QUEUE_MEMORY> | Pipeline queue memory limit per thread (default) or total | 768 |
--queue-memory-per-thread <QUEUE_MEMORY_PER_THREAD> | Interpret –queue-memory as per-thread (true, default) or total (false) | true |
--queue-memory-limit-mb <QUEUE_MEMORY_LIMIT_MB> | DEPRECATED: Use –queue-memory instead. Memory limit for pipeline queues in megabytes | |
--methylation-mode <METHYLATION_MODE> | Methylation-aware consensus calling mode. EM-Seq: C→T at ref-C = unmethylated (enzymatic conversion); TAPs: C→T at ref-C = methylated. Emits MM/ML methylation tags and cu/ct per-base count tags on consensus reads. Requires –ref | |
--ref <REFERENCE> | Path to the reference FASTA file (required when –methylation-mode is set) |
codec
Category: CONSENSUS
Call CODEC consensus reads from grouped BAM
Arguments
| Flag | Description | Default |
|---|---|---|
-i, --input <INPUT> | Input BAM file | required |
-o, --output <OUTPUT> | Output BAM file | required |
--async-reader <ASYNC_READER> | Enable async userspace prefetch on the input BAM | false |
-r, --rejects <REJECTS> | Optional output BAM file for rejected reads | |
-s, --stats <STATS> | Optional output file for statistics | |
-p, --read-name-prefix <READ_NAME_PREFIX> | Prefix for consensus read names | |
-R, --read-group-id <READ_GROUP_ID> | Read group ID for consensus reads | A |
-1, --error-rate-pre-umi <ERROR_RATE_PRE_UMI> | Phred-scaled error rate prior to UMI integration | 45 |
-2, --error-rate-post-umi <ERROR_RATE_POST_UMI> | Phred-scaled error rate post UMI integration | 40 |
-m, --min-input-base-quality <MIN_INPUT_BASE_QUALITY> | Minimum base quality in raw reads to use for consensus | 10 |
-B, --output-per-base-tags <OUTPUT_PER_BASE_TAGS> | Produce per-base tags (cd, ce) in addition to per-read tags | true |
--trim <TRIM> | Quality-trim reads before consensus calling (removes low-quality bases from ends) | false |
--min-consensus-base-quality <MIN_CONSENSUS_BASE_QUALITY> | Minimum consensus base quality (output consensus bases below this are masked to N) | 2 |
--threads <THREADS> | Number of threads for the multi-threaded pipeline | |
--compression-level <COMPRESSION_LEVEL> | Compression level for output BAM (0-12) | 1 |
-M, --min-reads <MIN_READS> | Minimum read pairs per strand to form consensus (same as –min-reads) | 1 |
--max-reads <MAX_READS> | Maximum read pairs per strand (downsample if exceeded) | |
-d, --min-duplex-length <MIN_DUPLEX_LENGTH> | Minimum duplex overlap length in bases | 1 |
--single-strand-qual <SINGLE_STRAND_QUAL> | Reduce single-strand region quality to this value (0-93). Note: This uses a different short flag than duplex’s -q for min-base-quality | |
-Q, --outer-bases-qual <OUTER_BASES_QUAL> | Reduce outer bases quality to this value (0-93) | |
-O, --outer-bases-length <OUTER_BASES_LENGTH> | Number of outer bases to reduce quality for | 5 |
-x, --max-duplex-disagreement-rate <MAX_DUPLEX_DISAGREEMENT_RATE> | Maximum duplex disagreement rate (0.0-1.0) | 1.0 |
-X, --max-duplex-disagreements <MAX_DUPLEX_DISAGREEMENTS> | Maximum number of duplex disagreements | |
--scheduler <SCHEDULER> | Scheduler strategy for thread work assignment | balanced-chase-drain |
--pipeline-stats <PIPELINE_STATS> | Print detailed pipeline statistics at completion | false |
--deadlock-timeout <DEADLOCK_TIMEOUT> | Timeout in seconds for deadlock detection (default: 10, 0 = disabled) | 10 |
--deadlock-recover <DEADLOCK_RECOVER> | Enable automatic deadlock recovery (default: false, detection only) | false |
--queue-memory <QUEUE_MEMORY> | Pipeline queue memory limit per thread (default) or total | 768 |
--queue-memory-per-thread <QUEUE_MEMORY_PER_THREAD> | Interpret –queue-memory as per-thread (true, default) or total (false) | true |
--queue-memory-limit-mb <QUEUE_MEMORY_LIMIT_MB> | DEPRECATED: Use –queue-memory instead. Memory limit for pipeline queues in megabytes |
filter
Category: POST-CONSENSUS
Filter consensus reads based on quality metrics
Description
Filters consensus reads generated by simplex or duplex commands. Two kinds of filtering are performed:
- Masking/filtering of individual bases in reads
- Filtering out of reads (i.e. not writing them to the output file)
Base-level filtering/masking is only applied if per-base tags are present (see duplex and simplex for descriptions of these tags). Read-level filtering is always applied. When filtering reads, secondary alignments and supplementary records may be removed independently if they fail one or more filters; if either R1 or R2 primary alignments fail a filter then all records for the template will be filtered out.
The filters applied are as follows:
- Reads with fewer than min-reads contributing reads are filtered out
- Reads with an average consensus error rate higher than max-read-error-rate are filtered out
- Reads with mean base quality of the consensus read, prior to any masking, less than min-mean-base-quality are filtered out (if specified)
- Bases with quality scores below min-base-quality are masked to Ns
- Bases with fewer than min-reads contributing raw reads are masked to Ns
- Bases with a consensus error rate (defined as the fraction of contributing reads that voted for a different base than the consensus call) higher than max-base-error-rate are masked to Ns
- Reads with a fraction or count of Ns higher than max-no-call-fraction after per-base filtering are filtered out.
When filtering single-umi consensus reads generated by simplex, a single value each should be supplied for –min-reads, –max-read-error-rate, and –max-base-error-rate.
When filtering duplex consensus reads generated by duplex, each of the three parameters may independently take 1-3 values. For example:
fgumi filter … –min-reads 10,5,3 –max-base-error-rate 0.1
In each case if fewer than three values are supplied, the last value is repeated (i.e. 80,40 -> 80 40 40
and 0.1 -> 0.1 0.1 0.1). The first value applies to the final consensus read, the second value to one
single-strand consensus, and the last value to the other single-strand consensus. It is required that if
values two and three differ, the more stringent value comes earlier.
In order to correctly filter reads in or out by template, the input BAM must be either queryname sorted or query grouped. If your BAM is not already in an appropriate order, this can be done in streaming fashion with:
fgumi sort -i in.bam –order queryname | fgumi filter -i /dev/stdin …
The output sort order may be specified with –sort-order. If not given, then the output will be in the same order as input.
The –reverse-per-base-tags option controls whether per-base tags should be reversed before being used on reads marked as being mapped to the negative strand. This is necessary if the reads have been mapped and the bases/quals reversed but the consensus tags have not. If true, the tags written to the output BAM will be reversed where necessary in order to line up with the bases and quals.
Arguments
| Flag | Description | Default |
|---|---|---|
-i, --input <INPUT> | Input BAM file | required |
-o, --output <OUTPUT> | Output BAM file | required |
--async-reader <ASYNC_READER> | Enable async userspace prefetch on the input BAM | false |
-r, --ref <REFERENCE> | Reference FASTA file for NM/UQ/MD tag regeneration. If not provided, alignment tag regeneration (NM/UQ/MD) is skipped | |
-M, --min-reads <MIN_READS> | Minimum number of raw reads to support a single-strand consensus base/read. For duplex: provide 1-3 values for [duplex, single-strand consensus, single-strand consensus] | |
-E, --max-read-error-rate <MAX_READ_ERROR_RATE> | Maximum raw read error rate for a single-strand consensus base/read (0.0-1.0). For duplex: provide 1-3 values for [duplex, single-strand consensus, single-strand consensus] | 0.025 |
-e, --max-base-error-rate <MAX_BASE_ERROR_RATE> | Maximum base error rate across raw reads (0.0-1.0). For duplex: provide 1-3 values for [duplex, AB consensus, BA consensus] | 0.1 |
-N, --min-base-quality <MIN_BASE_QUALITY> | Minimum base quality score (after masking) | |
-q, --min-mean-base-quality <MIN_MEAN_BASE_QUALITY> | Minimum mean base quality across the read (after masking) | |
-n, --max-no-call-fraction <MAX_NO_CALL_FRACTION> | Maximum no-calls (N bases) allowed in a read | 0.2 |
-R, --reverse-per-base-tags <REVERSE_PER_BASE_TAGS> | Reverse per-base tags for negative strand reads | false |
--threads <THREADS> | Number of threads for the multi-threaded pipeline | |
--filter-by-template <FILTER_BY_TEMPLATE> | Filter templates together (all primary reads must pass) | true |
--rejects <REJECTS> | Optional output BAM file for rejected reads | |
--stats <STATS> | Optional output file for filtering statistics | |
-s, --require-single-strand-agreement <REQUIRE_SINGLE_STRAND_AGREEMENT> | Require single-strand agreement for duplex consensus (mask bases where AB and BA disagree) | false |
--min-methylation-depth <MIN_METHYLATION_DEPTH> | Minimum methylation depth (cu+ct) to keep a base call (EM-Seq/TAPs). For duplex: provide 1-3 values for [duplex, AB consensus, BA consensus] | |
--require-strand-methylation-agreement <REQUIRE_STRAND_METHYLATION_AGREEMENT> | Require strand methylation agreement at CpG sites for duplex consensus (EM-Seq/TAPs). Masks both positions of a CpG dinucleotide when top and bottom strands disagree on methylation status. Requires –ref | false |
--min-conversion-fraction <MIN_CONVERSION_FRACTION> | Minimum bisulfite/enzymatic conversion fraction at non-CpG cytosines. For EM-Seq: checks converted/total >= threshold (high conversion = good). For TAPs: checks unconverted/total >= threshold (low conversion = good). Requires –ref and –methylation-mode. Uses cu/ct tags | |
--methylation-mode <METHYLATION_MODE> | Methylation mode for conversion fraction filtering. Required when using –min-conversion-fraction. Controls whether the conversion fraction check uses converted (em-seq) or unconverted (taps) counts as the numerator | |
--compression-level <COMPRESSION_LEVEL> | Compression level for output BAM (0-12) | 1 |
--scheduler <SCHEDULER> | Scheduler strategy for thread work assignment | balanced-chase-drain |
--pipeline-stats <PIPELINE_STATS> | Print detailed pipeline statistics at completion | false |
--deadlock-timeout <DEADLOCK_TIMEOUT> | Timeout in seconds for deadlock detection (default: 10, 0 = disabled) | 10 |
--deadlock-recover <DEADLOCK_RECOVER> | Enable automatic deadlock recovery (default: false, detection only) | false |
--queue-memory <QUEUE_MEMORY> | Pipeline queue memory limit per thread (default) or total | 768 |
--queue-memory-per-thread <QUEUE_MEMORY_PER_THREAD> | Interpret –queue-memory as per-thread (true, default) or total (false) | true |
--queue-memory-limit-mb <QUEUE_MEMORY_LIMIT_MB> | DEPRECATED: Use –queue-memory instead. Memory limit for pipeline queues in megabytes |
clip
Category: POST-CONSENSUS
Clip overlapping reads in BAM files
Description
Clips reads from the same template. Ensures that at least N bases are clipped from any end of the read (i.e. R1 5’ end, R1 3’ end, R2 5’ end, and R2 3’ end). Optionally clips reads from the same template to eliminate overlap between the reads. This ensures that downstream processes, particularly variant calling, cannot double-count evidence from the same template when both reads span a variant site in the same template.
Clipping overlapping reads is only performed on FR read pairs, and is implemented by clipping approximately half the overlapping bases from each read. By default soft clipping is performed.
Secondary alignments and supplemental alignments are not clipped, but are passed through into the output.
In order to correctly clip reads by template and update mate information, the input BAM must be either queryname sorted or query grouped. If your input BAM is not in an appropriate order the sort can be done in streaming fashion with, for example:
fgumi sort -i in.bam –order queryname | fgumi clip -i /dev/stdin …
The output sort order may be specified with –sort-order. If not given, then the output will be in the same order as input.
Any existing NM, UQ and MD tags are repaired, and mate-pair information is updated.
Three clipping modes are supported:
soft- soft-clip the bases and qualities.soft-with-mask- soft-clip and mask the bases and qualities (make bases Ns and qualities the minimum).hard- hard-clip the bases and qualities.
The –upgrade-clipping parameter will convert all existing clipping in the input to the given more stringent mode:
from soft to either soft-with-mask or hard, and soft-with-mask to hard. In all other cases, clipping remains
the same prior to applying any other clipping criteria.
Arguments
| Flag | Description | Default |
|---|---|---|
-i, --input <INPUT> | Input BAM file | required |
-o, --output <OUTPUT> | Output BAM file | required |
--async-reader <ASYNC_READER> | Enable async userspace prefetch on the input BAM | false |
-r, --reference <REFERENCE> | Reference FASTA file (required for tag regeneration) | required |
-c, --clipping-mode <CLIPPING_MODE> | Clipping mode: soft, soft-with-mask, or hard | hard |
-S, --sort-order <SORT_ORDER> | Output sort order (if not specified, output is in same order as input) | |
--clip-overlapping-reads <CLIP_OVERLAPPING_READS> | Clip overlapping read pairs | false |
--clip-bases-past-mate <CLIP_EXTENDING_PAST_MATE> | Clip reads that extend past their mate’s start position | false |
--read-one-five-prime <READ_ONE_FIVE_PRIME> | Minimum bases to clip from 5’ end of R1 | 0 |
--read-one-three-prime <READ_ONE_THREE_PRIME> | Minimum bases to clip from 3’ end of R1 | 0 |
--read-two-five-prime <READ_TWO_FIVE_PRIME> | Minimum bases to clip from 5’ end of R2 | 0 |
--read-two-three-prime <READ_TWO_THREE_PRIME> | Minimum bases to clip from 3’ end of R2 | 0 |
-H, --upgrade-clipping <UPGRADE_CLIPPING> | Upgrade existing clipping to the specified clipping mode | false |
-a, --auto-clip-attributes <AUTO_CLIP_ATTRIBUTES> | Automatically clip extended attributes that match read length | false |
-m, --metrics <METRICS> | Output file for clipping metrics | |
--threads <THREADS> | Number of threads for the multi-threaded pipeline | |
--compression-level <COMPRESSION_LEVEL> | Compression level for output BAM (0-12) | 1 |
--scheduler <SCHEDULER> | Scheduler strategy for thread work assignment | balanced-chase-drain |
--pipeline-stats <PIPELINE_STATS> | Print detailed pipeline statistics at completion | false |
--deadlock-timeout <DEADLOCK_TIMEOUT> | Timeout in seconds for deadlock detection (default: 10, 0 = disabled) | 10 |
--deadlock-recover <DEADLOCK_RECOVER> | Enable automatic deadlock recovery (default: false, detection only) | false |
--queue-memory <QUEUE_MEMORY> | Pipeline queue memory limit per thread (default) or total | 768 |
--queue-memory-per-thread <QUEUE_MEMORY_PER_THREAD> | Interpret –queue-memory as per-thread (true, default) or total (false) | true |
--queue-memory-limit-mb <QUEUE_MEMORY_LIMIT_MB> | DEPRECATED: Use –queue-memory instead. Memory limit for pipeline queues in megabytes |
duplex-metrics
Category: POST-CONSENSUS
Collect QC metrics for duplex consensus reads
Description
Collects a suite of metrics to QC duplex sequencing data.
Inputs
The input to this tool must be a BAM file that is either:
- The exact BAM output by the
grouptool (in the sort-order it was produced in) - A BAM file that has MI tags present on all reads (usually set by
groupand has been sorted into template-coordinate order
Calculation of metrics may be restricted to a set of regions using the --intervals parameter.
This can significantly affect results as off-target reads in duplex sequencing experiments often
have very different properties than on-target reads due to the lack of enrichment.
Several metrics are calculated related to the fraction of tag families that have duplex coverage.
The definition of “duplex” is controlled by the --min-ab-reads and --min-ba-reads parameters.
The default is to treat any tag family with at least one observation of each strand as a duplex,
but this could be made more stringent, e.g. by setting --min-ab-reads=3 --min-ba-reads=3.
Outputs
The following output files are produced:
- <output>.family_sizes.txt: metrics on the frequency of different types of families of different sizes
- <output>.duplex_family_sizes.txt: metrics on the frequency of duplex tag families by the number of observations from each strand
- <output>.duplex_yield_metrics.txt: summary QC metrics produced using 5%, 10%, 15%…100% of the data
- <output>.umi_counts.txt: metrics on the frequency of observations of UMIs within reads and tag families
- <output>.duplex_umi_counts.txt: (optional) metrics on the frequency of observations of duplex UMIs within
reads and tag families. This file is only produced if the
--duplex-umi-countsoption is used as it requires significantly more memory to track all pairs of UMIs seen when a large number of UMI sequences are present. - <output>.duplex_qc.pdf: (optional) a series of plots generated from the preceding metrics files for
visualization. This file is only produced if R is available with the required
packages (ggplot2 and scales). Use
--descriptionto customize plot titles.
Within the metrics files the prefixes CS, SS and DS are used to mean:
- CS: tag families where membership is defined solely on matching genome coordinates and strand
- SS: single-stranded tag families where membership is defined by genome coordinates, strand and UMI; ie. 50/A and 50/B are considered different tag families
- DS: double-stranded tag families where membership is collapsed across single-stranded tag families from the same double-stranded source molecule; i.e. 50/A and 50/B become one family
Arguments
| Flag | Description | Default |
|---|---|---|
-i, --input <INPUT> | Input BAM file (UMI-grouped, from group) | required |
-o, --output <OUTPUT> | Output prefix for metrics files | required |
--min-ab-reads <MIN_AB_READS> | Minimum AB reads to call a duplex | 1 |
--min-ba-reads <MIN_BA_READS> | Minimum BA reads to call a duplex | 1 |
--duplex-umi-counts <DUPLEX_UMI_COUNTS> | Collect duplex UMI counts (memory intensive) | false |
-l, --intervals <INTERVALS> | Optional intervals file to restrict analysis (BED or Picard interval list format) | |
--description <DESCRIPTION> | Optional sample name or description for PDF plot titles |
review
Category: POST-CONSENSUS
Extract data to review variant calls from consensus reads
Description
Extracts data to make reviewing of variant calls from consensus reads easier.
Creates a list of variant sites from the input VCF (SNPs only) or IntervalList then extracts all the consensus reads that do not contain a reference allele at the variant sites, and all raw reads that contributed to those consensus reads. This will include consensus reads that carry the alternate allele, a third allele, a no-call or a spanning deletion at the variant site.
Reads are correlated between consensus and grouped BAMs using a molecule ID stored in an optional
attribute, MI by default. In order to support paired molecule IDs where two or more molecule IDs
are related (e.g. see the Paired assignment strategy in group) the molecule ID is truncated at
the last / if present (e.g. 1/A => 1 and 2 => 2).
Both input BAMs must be coordinate sorted and indexed.
Output Files
A pair of output BAMs are created:
- <output>.consensus.bam: Contains the relevant consensus reads from the consensus BAM
- <output>.grouped.bam: Contains the relevant raw reads from the grouped BAM
A review file <output>.txt is also created. The review file contains details on each variant
position along with detailed information on each consensus read that supports the variant. If the
--sample argument is supplied and the input is VCF, genotype information for that sample will be
retrieved. If the sample name isn’t supplied and the VCF contains only a single sample then those
genotypes will be used.
The --maf parameter controls which variants get detailed per-read information in the output file.
Only variants with a minor allele frequency at or below this threshold will have detailed information
written.
Arguments
| Flag | Description | Default |
|---|---|---|
-i, --input <INPUT> | Input VCF or IntervalList of variant locations | required |
-c, --consensus-bam <CONSENSUS_BAM> | BAM file of consensus reads used to call variants | required |
-g, --grouped-bam <GROUPED_BAM> | BAM file of grouped raw reads used to build consensuses | required |
-r, --ref <REFERENCE> | Reference FASTA file | required |
-o, --output <OUTPUT> | Output prefix for generated files | required |
-s, --sample <SAMPLE> | Name of sample being reviewed (for VCF genotype extraction) | |
-N, --ignore-ns <IGNORE_NS> | Ignore N bases in consensus reads | false |
-m, --maf <MAF> | Only output detailed information for variants at or below this MAF | 0.05 |
simplex-metrics
Category: POST-CONSENSUS
Collect QC metrics for simplex sequencing data
Description
Collects a suite of metrics to QC simplex sequencing data.
Inputs
The input to this tool must be a BAM file that is either:
- The exact BAM output by the
grouptool (in the sort-order it was produced in) - A BAM file that has MI tags present on all reads (usually set by
groupand has been sorted into template-coordinate order
Calculation of metrics may be restricted to a set of regions using the --intervals parameter.
This can significantly affect results as off-target reads often have very different properties
than on-target reads due to the lack of enrichment.
Outputs
The following output files are produced:
- <output>.family_sizes.txt: metrics on the frequency of CS and SS families of different sizes
- <output>.simplex_yield_metrics.txt: summary QC metrics produced using 5%, 10%, 15%…100% of the data
- <output>.umi_counts.txt: metrics on the frequency of observations of UMIs within reads and tag families
- <output>.simplex_qc.pdf: (optional) a series of plots generated from the preceding metrics files for
visualization. This file is only produced if R is available with the required
packages (ggplot2 and scales). Use
--descriptionto customize plot titles.
Within the metrics files the prefixes CS and SS are used to mean:
- CS: tag families where membership is defined solely on matching genome coordinates and strand
- SS: single-stranded tag families where membership is defined by genome coordinates, strand and UMI
Arguments
| Flag | Description | Default |
|---|---|---|
-i, --input <INPUT> | Input BAM file (UMI-grouped, from group) | required |
-o, --output <OUTPUT> | Output prefix for metrics files | required |
--min-reads <MIN_READS> | Minimum reads per SS family to count as a consensus family in yield metrics | 1 |
-l, --intervals <INTERVALS> | Optional intervals file to restrict analysis (BED or Picard interval list format) | |
--description <DESCRIPTION> | Optional sample name or description for PDF plot titles |
downsample
Category: UTILITIES
Downsample BAM by UMI family using streaming
Description
Downsample a BAM file by UMI family using a single-pass streaming algorithm.
This tool reads a BAM file that has been processed by fgumi group (or fgbio GroupReadsByUmi) containing MI tags, uniformly samples UMI families, and outputs kept reads directly to a BAM file.
Requires input BAM to be in template-coordinate order:
- SO:unsorted (or not set)
- GO:query
- SS:unsorted:template-coordinate or SS:template-coordinate
The tool processes families in streaming fashion by grouping consecutive reads with the same MI tag value. For each family, a random decision is made based on the fraction parameter to either keep or reject all reads in that family.
Example usage: fgumi downsample -i grouped.bam -o downsampled.bam -f 0.1 –seed 42 fgumi downsample -i grouped.bam -o kept.bam -f 0.5 –rejects rejected.bam fgumi downsample -i grouped.bam -o kept.bam -f 0.1 –histogram-kept kept_hist.txt
Arguments
| Flag | Description | Default |
|---|---|---|
-i, --input <INPUT> | Input BAM file | required |
-o, --output <OUTPUT> | Output BAM file | required |
--async-reader <ASYNC_READER> | Enable async userspace prefetch on the input BAM | false |
-f, --fraction <FRACTION> | Fraction of UMI families to keep (0.0 exclusive to 1.0 inclusive) | required |
--rejects <REJECTS> | Optional output BAM file for rejected reads | |
--seed <SEED> | Random seed for reproducibility | |
--validate-mi-order <VALIDATE_MI_ORDER> | Validate that MI tags appear in consecutive groups (error if seen non-consecutively) | false |
--histogram-kept <HISTOGRAM_KEPT> | Output file for kept family size histogram | |
--histogram-rejected <HISTOGRAM_REJECTED> | Output file for rejected family size histogram | |
--compression-level <COMPRESSION_LEVEL> | Compression level for output BAM (0-12) | 1 |
Metrics Reference
Auto-generated from fgumi metric struct definitions.
| Metric | Description |
|---|---|
| FamilySizeMetric | Metrics quantifying the distribution of different kinds of read family sizes. |
| DuplexFamilySizeMetric | Metrics describing double-stranded (duplex) tag families by AB and BA strand sizes. |
| DuplexYieldMetric | Metrics sampled at various levels of coverage via random downsampling. |
| DuplexUmiMetric | Metrics describing observed duplex UMI sequences and their frequencies. |
| UmiMetric | Metrics describing observed UMI sequences and their observation frequencies. |
| UmiCorrectionMetrics | Metrics tracking how well observed UMIs match expected UMI sequences. |
| ClippingMetrics | Clipping metrics for a specific read type |
| UmiGroupingMetrics | Metrics for UMI grouping operations. |
| FamilySizeMetrics | Family size distribution metrics. |
| PositionGroupSizeMetrics | Position group size distribution metrics. |
| SimplexFamilySizeMetric | Metrics quantifying the distribution of CS and SS read family sizes. |
| SimplexYieldMetric | Metrics sampled at various levels of coverage via random downsampling for simplex experiments. |
| ConsensusMetrics | Consensus calling metrics with rejection tracking. |
UmiMetric
Metrics describing observed UMI sequences and their observation frequencies.
UMI sequences may be corrected using information within a double-stranded tag family.
Fields
| Column | Type | Description |
|---|---|---|
umi | String | The UMI sequence (possibly corrected) |
raw_observations | usize | Number of read pairs observing this UMI (after correction) |
raw_observations_with_errors | usize | Subset of raw observations that underwent correction |
unique_observations | usize | Number of double-stranded tag families observing this UMI |
fraction_raw_observations | f64 | Fraction of all raw observations |
fraction_unique_observations | f64 | Fraction of all unique observations |
UmiCorrectionMetrics
Metrics tracking how well observed UMIs match expected UMI sequences.
These metrics are generated per-UMI and track the distribution of match types (perfect matches, single mismatches, etc.) for each expected UMI.
Fields
umi- The expected/corrected UMI sequence (or all Ns for unmatched)total_matches- Total UMI sequences matched/corrected to this UMIperfect_matches- Number of reads with zero mismatchesone_mismatch_matches- Number of reads with exactly one mismatchtwo_mismatch_matches- Number of reads with exactly two mismatchesother_matches- Number of reads with three or more mismatchesfraction_of_matches- Proportion of all reads matching this UMIrepresentation- Ratio of this UMI’s count to the mean count across all UMIs
Fields
| Column | Type | Description |
|---|---|---|
umi | String | The corrected UMI sequence (or all Ns for unmatched). |
total_matches | u64 | The number of UMI sequences that matched/were corrected to this UMI. |
perfect_matches | u64 | The number of UMI sequences that were perfect matches to this UMI. |
one_mismatch_matches | u64 | The number of UMI sequences that matched with a single mismatch. |
two_mismatch_matches | u64 | The number of UMI sequences that matched with two mismatches. |
other_matches | u64 | The number of UMI sequences that matched with three or more mismatches. |
fraction_of_matches | f64 | The fraction of all UMIs that matched or were corrected to this UMI. |
representation | f64 | The total_matches for this UMI divided by the mean total_matches for all UMIs. |
UmiGroupingMetrics
Metrics for UMI grouping operations.
These metrics track how reads are grouped by UMI and provide insight into data quality and molecule representation.
Fields
| Column | Type | Description |
|---|---|---|
total_records | u64 | Total SAM records processed |
accepted_records | u64 | Records accepted for grouping |
discarded_non_pf | u64 | Records discarded (not passing filter) |
discarded_poor_alignment | u64 | Records discarded (poor alignment quality) |
discarded_ns_in_umi | u64 | Records discarded (Ns in UMI) |
discarded_umi_too_short | u64 | Records discarded (UMI too short) |
unique_molecule_ids | u64 | Number of unique molecule IDs assigned |
total_families | u64 | Total number of UMI families/groups |
avg_reads_per_molecule | f64 | Average reads per molecule |
median_reads_per_molecule | u64 | Median reads per molecule |
min_reads_per_molecule | u64 | Minimum reads per molecule |
max_reads_per_molecule | u64 | Maximum reads per molecule |
FamilySizeMetric
Metrics quantifying the distribution of different kinds of read family sizes.
Three kinds of families are described:
- CS (Coordinate & Strand): families grouped by unclipped 5’ genomic positions and strands
- SS (Single Strand): single-strand families using UMIs, not linking opposing strands
- DS (Double Strand): families combining single-strand families from opposite strands
Fields
| Column | Type | Description |
|---|---|---|
family_size | usize | The family size (number of read pairs grouped together) |
cs_count | usize | Count of CS families with this size |
cs_fraction | f64 | Fraction of all CS families with this size |
cs_fraction_gt_or_eq_size | f64 | Fraction of CS families with size >= family_size |
ss_count | usize | Count of SS families with this size |
ss_fraction | f64 | Fraction of all SS families with this size |
ss_fraction_gt_or_eq_size | f64 | Fraction of SS families with size >= family_size |
ds_count | usize | Count of DS families with this size |
ds_fraction | f64 | Fraction of all DS families with this size |
ds_fraction_gt_or_eq_size | f64 | Fraction of DS families with size >= family_size |
FamilySizeMetrics
Family size distribution metrics.
Describes the distribution of UMI family sizes in the dataset.
Fields
| Column | Type | Description |
|---|---|---|
family_size | usize | Family size (number of reads) |
count | u64 | Number of families with this size |
fraction | f64 | Fraction of all families with this size |
fraction_gt_or_eq_family_size | f64 | Cumulative fraction (families with size >= this value) |
PositionGroupSizeMetrics
Position group size distribution metrics.
Describes the distribution of position group sizes (the number of unique UMI families sharing the same start/end coordinates) in the dataset.
Fields
| Column | Type | Description |
|---|---|---|
position_group_size | usize | Position group size (number of unique UMI families at the same genomic position) |
count | u64 | Number of position groups with this size |
fraction | f64 | Fraction of all position groups with this size |
fraction_gt_or_eq_position_group_size | f64 | Cumulative fraction (position groups with size >= this value) |
DuplexFamilySizeMetric
Metrics describing double-stranded (duplex) tag families by AB and BA strand sizes.
For a given tag family, ab is the larger sub-family and ba is the smaller one.
Fields
| Column | Type | Description |
|---|---|---|
ab_size | usize | Number of reads in the AB sub-family (larger) |
ba_size | usize | Number of reads in the BA sub-family (smaller) |
count | usize | Count of families with these AB/BA sizes |
fraction | f64 | Fraction of all duplex families with these sizes |
fraction_gt_or_eq_size | f64 | Fraction of duplex families with AB >= ab_size and BA >= ba_size |
DuplexYieldMetric
Metrics sampled at various levels of coverage via random downsampling.
Fields
| Column | Type | Description |
|---|---|---|
fraction | f64 | Approximate fraction of full dataset used |
read_pairs | usize | Number of read pairs upon which metrics are based |
cs_families | usize | Number of CS (Coordinate & Strand) families |
ss_families | usize | Number of SS (Single-Strand by UMI) families |
ds_families | usize | Number of DS (Double-Strand by UMI) families |
ds_duplexes | usize | Number of DS families that are duplexes (min reads on both strands) |
ds_fraction_duplexes | f64 | Fraction of DS families that are duplexes |
ds_fraction_duplexes_ideal | f64 | Expected fraction of DS families that should be duplexes under ideal model |
DuplexUmiMetric
Metrics describing observed duplex UMI sequences and their frequencies.
Duplex UMIs are normalized to F1R2 orientation (positive strand first).
Fields
| Column | Type | Description |
|---|---|---|
umi | String | The duplex UMI sequence (possibly corrected, F1R2 normalized) |
raw_observations | usize | Number of read pairs observing this duplex UMI |
raw_observations_with_errors | usize | Subset of raw observations that underwent correction |
unique_observations | usize | Number of double-stranded tag families observing this duplex UMI |
fraction_raw_observations | f64 | Fraction of all raw observations |
fraction_unique_observations | f64 | Fraction of all unique observations |
fraction_unique_observations_expected | f64 | Expected fraction based on individual UMI frequencies |
SimplexFamilySizeMetric
Metrics quantifying the distribution of CS and SS read family sizes.
Two kinds of families are described:
- CS (Coordinate & Strand): families grouped by unclipped 5’ genomic positions and strands
- SS (Single Strand): single-strand families using UMIs, not linking opposing strands
Fields
| Column | Type | Description |
|---|---|---|
family_size | usize | The family size (number of read pairs grouped together) |
cs_count | usize | Count of CS families with this size |
cs_fraction | f64 | Fraction of all CS families with this size |
cs_fraction_gt_or_eq_size | f64 | Fraction of CS families with size >= family_size |
ss_count | usize | Count of SS families with this size |
ss_fraction | f64 | Fraction of all SS families with this size |
ss_fraction_gt_or_eq_size | f64 | Fraction of SS families with size >= family_size |
SimplexYieldMetric
Metrics sampled at various levels of coverage via random downsampling for simplex experiments.
Fields
| Column | Type | Description |
|---|---|---|
fraction | f64 | Approximate fraction of full dataset used |
read_pairs | usize | Number of read pairs upon which metrics are based |
cs_families | usize | Number of CS (Coordinate & Strand) families |
ss_families | usize | Number of SS (Single-Strand by UMI) families |
mean_ss_family_size | f64 | Mean SS family size |
ss_singletons | usize | Number of SS singleton families (size 1) |
ss_singleton_fraction | f64 | Fraction of SS families that are singletons |
ss_consensus_families | usize | Number of SS families with size >= consensus minimum |
ConsensusMetrics
Consensus calling metrics with rejection tracking.
These metrics track the consensus calling process, including how many reads were accepted, filtered, and the reasons for rejection.
Fields
| Column | Type | Description |
|---|---|---|
total_input_reads | u64 | Total input reads processed |
consensus_reads | u64 | Number of consensus reads generated |
filtered_reads | u64 | Number of input reads filtered out |
total_umi_groups | u64 | Total number of UMI groups processed |
umi_groups_with_consensus | u64 | UMI groups that generated consensus |
umi_groups_failed | u64 | UMI groups that failed to generate consensus |
avg_input_reads_per_consensus | f64 | Average input reads per consensus read |
avg_raw_read_depth | f64 | Average raw read depth per consensus read |
min_raw_read_depth | u64 | Minimum raw read depth |
max_raw_read_depth | u64 | Maximum raw read depth |
rejected_insufficient_support | u64 | Reads rejected due to insufficient support |
rejected_minority_alignment | u64 | Reads rejected due to minority alignment |
rejected_insufficient_strand_support | u64 | Reads rejected due to insufficient strand support |
rejected_low_base_quality | u64 | Reads rejected due to low base quality |
rejected_excessive_n_bases | u64 | Reads rejected due to excessive N bases |
rejected_no_valid_alignment | u64 | Reads rejected due to no valid alignment |
rejected_low_mapping_quality | u64 | Reads rejected due to low mapping quality |
rejected_n_bases_in_umi | u64 | Reads rejected due to N bases in UMI |
rejected_missing_umi | u64 | Reads rejected due to missing UMI tag |
rejected_not_passing_filter | u64 | Reads rejected due to not passing filter |
rejected_low_mean_quality | u64 | Reads rejected due to low mean quality |
rejected_insufficient_min_depth | u64 | Reads rejected due to insufficient min depth |
rejected_excessive_error_rate | u64 | Reads rejected due to excessive error rate |
rejected_umi_too_short | u64 | Reads rejected due to UMI too short |
rejected_same_strand_only | u64 | Reads rejected due to same strand only |
rejected_duplicate_umi | u64 | Reads rejected due to duplicate UMI |
rejected_orphan_consensus | u64 | Reads rejected due to orphan consensus (only R1 or R2 had consensus) |
rejected_zero_bases_post_trimming | u64 | Reads rejected due to zero bases after trimming |
ClippingMetrics
Clipping metrics for a specific read type
Fields
| Column | Type | Description |
|---|---|---|
read_type | ReadType | The type of read this metric applies to |
reads | usize | Total number of reads examined |
reads_unmapped | usize | Number of reads that became unmapped due to clipping |
reads_clipped_pre | usize | Number of reads with any clipping before clip |
reads_clipped_post | usize | Number of reads with any clipping after clip |
reads_clipped_five_prime | usize | Number of reads clipped on 5’ end |
reads_clipped_three_prime | usize | Number of reads clipped on 3’ end |
reads_clipped_overlapping | usize | Number of reads clipped due to overlapping reads |
reads_clipped_extending | usize | Number of reads clipped due to extending past mate |
bases | usize | Total number of aligned bases after clipping |
bases_clipped_pre | usize | Number of bases clipped before clip |
bases_clipped_post | usize | Number of bases clipped after clip |
bases_clipped_five_prime | usize | Number of bases clipped on 5’ end |
bases_clipped_three_prime | usize | Number of bases clipped on 3’ end |
bases_clipped_overlapping | usize | Number of bases clipped due to overlapping reads |
bases_clipped_extending | usize | Number of bases clipped due to extending past mate |