fgumi
High-performance tools for UMI-tagged sequencing data: extraction, grouping, and consensus calling.
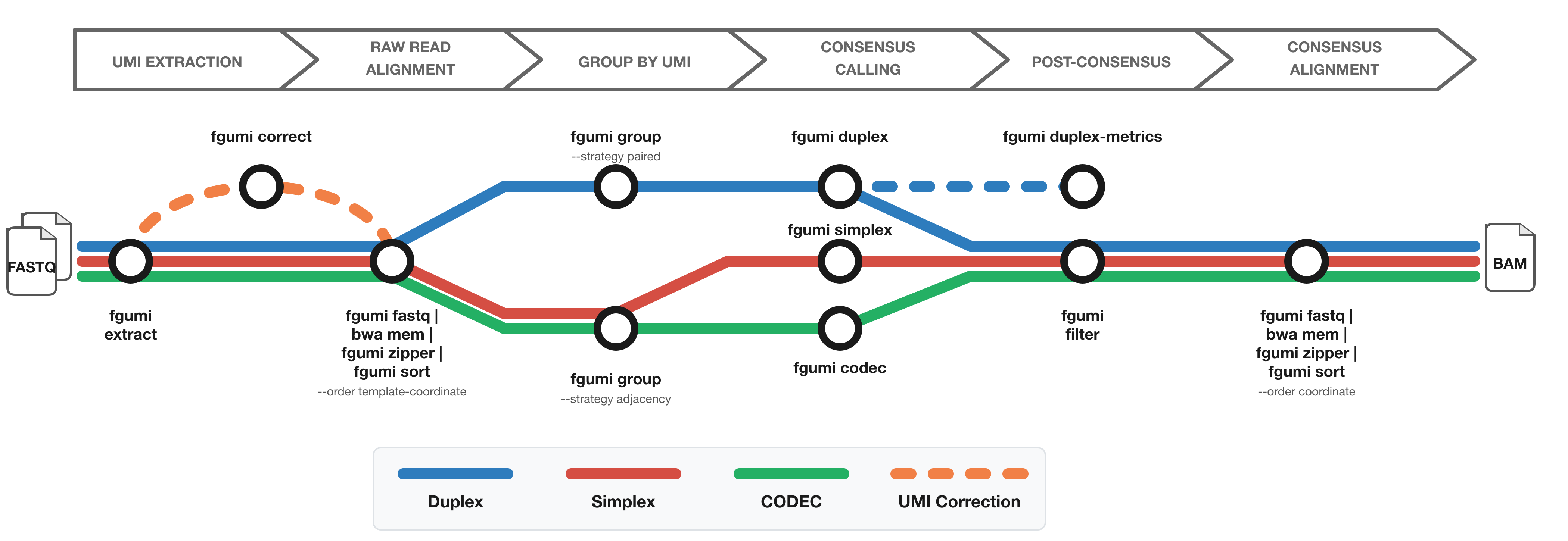
The diagram shows the workflow from FASTQ files to filtered consensus reads:
- Red: Simplex (single-strand) consensus
- Blue: Duplex (double-strand) consensus
- Green: CODEC consensus
- Orange: Optional UMI correction for fixed UMI sets
Where to Use fgumi
Command Line
Install and run fgumi directly on your data. See the Getting Started guide.
Nextflow Pipeline
Use fastquorum for an end-to-end Nextflow workflow from FASTQ to consensus reads using fgumi.
Latch.bio
Run fgumi in the cloud with a point-and-click interface via Latch.bio — no installation required.
Installation
Pre-built Binaries
Pre-built binaries for common operating systems and architectures are attached to each release.
Cargo
cargo install fgumi
Bioconda
conda install -c bioconda fgumi
From Source
git clone https://github.com/fulcrumgenomics/fgumi
cd fgumi
cargo build --release
Available Commands
| Command | Description |
|---|---|
extract | Extract UMIs from FASTQ files |
correct | Correct UMIs based on sequence similarity |
fastq | Convert BAM to FASTQ format |
zipper | Restore original FASTQ from unaligned BAM |
sort | Sort BAM by coordinate/queryname/template |
group | Group reads by UMI |
dedup | Mark/remove UMI-aware duplicates |
simplex | Call single-strand consensus reads |
duplex | Call duplex consensus reads |
codec | Call CODEC consensus |
filter | Filter consensus reads |
clip | Clip overlapping read pairs |
duplex-metrics | Collect duplex metrics |
review | Review consensus variants |
downsample | Downsample BAM by UMI family |
simplex-metrics | Collect simplex metrics |
merge | Merge sorted BAM files |
See the Tool Reference for detailed documentation of each command.